
| Aldehyde |
| 👁 Allgemeine Struktur eines Aldehyds |
| Allgemeine Struktur eines Aldehyds. Der Rest R kann ein Wasserstoffatom oder ein Organyl-Rest (Alkyl-, Aryl-, Alkenyl-Rest etc.) sein. Die Aldehydgruppe (Formylgruppe) ist blau gekennzeichnet. |
| 👁 Beispiele |
| Beispiele: Formaldehyd (Methanal, links), Acetaldehyd (Ethanal, Mitte) und Propionaldehyd (Propanal, rechts) mit blau gekennzeichneter Aldehydgruppe (Formylgruppe) |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Aldehyde (aus neulateinisch alcoholus dehydrogenatus, „dehydrierter Alkohol“ oder „Alkohol, dem Wasserstoff entzogen wurde“) sind chemische Verbindungen mit der funktionellen Gruppe –CHO, die Aldehydgruppe oder auch Formylgruppe genannt wird. Die Carbonylgruppe der Aldehyde trägt im Unterschied zu den Ketonen einen Wasserstoff- und einen Kohlenstoffsubstituenten. Eine Ausnahme bildet der einfachste Aldehyd Methanal (Formaldehyd), der zwei Wasserstoffsubstituenten trägt. Aldehyde mit einem Alkylrest (also Alkan-Derivate) werden als Alkanale bezeichnet; deren homologe Reihe leitet sich nomenklatorisch entsprechend von der homologen Reihe der Alkane ab. Weiter existieren Mehrfachaldehyde – wie beispielsweise das Glyoxal, der einfachste Dialdehyd.
Geschichte
[Bearbeiten | Quelltext bearbeiten]Acetaldehyd wurde zum ersten Mal 1774 von Carl Wilhelm Scheele durch Oxidation von Ethanol mit Mangandioxid hergestellt. 1835 stellte Justus von Liebig Acetaldehyd durch Oxidation von Ethanol mit Chromsäure her und führte den Stoffgruppennamen Aldehyde, abgeleitet von alcoholus dehydrogenatus, ein.[1] Liebig fand heraus, dass Acetaldehyd eine Zwischenstufe in der Reaktion von Ethanol zu Essigsäure ist und entdeckte die Silberspiegelprobe, mit der Aldehyde nachgewiesen werden können.[2]
Einige Reaktionen der Aldehyde sind schon sehr lange bekannt. In den 1860er-Jahren beschrieb Hugo Schiff die Herstellung von Iminen aus Aldehyden und primären Aminen, die deshalb auch Schiff-Basen genannt werden. Zu dieser Zeit kamen gerade erst die ersten grundlegenden Erkenntnisse zur Struktur organischer Moleküle auf, beispielsweise die Vierbindigkeit des Kohlenstoffs und die Struktur des Benzolrings.[3][4]
Nomenklatur
[Bearbeiten | Quelltext bearbeiten]Im einfachsten Fall erhalten Aldehyde nach der IUPAC-Nomenklatur den Namen des Alkans mit dem Suffix -al. Dementsprechend heißt der vom Methan abgeleitete Aldehyd Methanal, der vom Ethan abgeleitete Ethanal.[5] Da die Aldehydfunktion nur am Kettenanfang stehen kann, ist es nicht nötig, mit einem Lokanten seine Position anzugeben.[6] In der Prioritätenrangfolge der funktionellen Gruppen kommen Aldehyde nach den Carbonsäuren und deren Derivaten (z. B. Ester, Amide und Nitrile), aber vor den Ketonen. Kommt eine ranghöhere funktionelle Gruppe im Molekül vor, muss die Aldehydfunktion mit einem Präfix benannt werden. Ist sie Teil des Stammsystems, wird das Präfix „Oxo-“ verwendet, z. B. 3-Oxopropansäure. Andernfalls wird das Präfix „Formyl-“ verwendet, das für die Aldehydgruppe inklusive des Kohlenstoffatoms gilt, z. B. 4-Formylbenzoesäure.[7] Ist die Aldehydfunktion die funktionelle Gruppe mit der höchsten Priorität, wird aber nicht zum Stammsystem gerechnet, weil sie beispielsweise an einen Ring gebunden ist, dann muss analog zur Endung „-carbonsäure“ bei Carbonsäuren die Endung „-carbaldehyd“ angehängt werden. Dabei wird durch die Endung ebenfalls die komplette Formylgruppe beschrieben, während bei der Endung „-al“ das Kohlenstoffatom dem Stammsystem zugerechnet wird.[8]
Der Trivialname leitet sich von der lateinischen Bezeichnung für die Carbonsäure der gleichen Kettenlänge her. Für das Methanal (H–CHO) ist das die Methansäure (lat. acidum formicum, H–COOH), daher Formaldehyd, für das Ethanal die Ethansäure (lat. acidum aceticum, CH3–COOH), daher Acetaldehyd. Die Trivialnamen der einzelnen Aldehyde sind grammatikalisch oft maskulin, z. B. der Acetaldehyd, aber laut Duden auch sächlich, also das Acetaldehyd. Dagegen sind die Vertreter der Alkanale immer Neutra, z. B. das Ethanal. Entsprechend leiten sich die anderen Trivialnamen ab. Dicarbonsäuren, bei denen eine Carbonsäuregruppe zu einer Aldehydgruppe reduziert wurde, werden gelegentlich Semialdehyde genannt.[9]
Abgrenzung zu verwandten Verbindungen
[Bearbeiten | Quelltext bearbeiten]Der einfachste Aldehyd ist Formaldehyd, bei dem zwei Wasserstoffatome an die Carbonylgruppe gebunden sind. Bei anderen Aldehyden sind an die Carbonylgruppe ein Wasserstoffatom und ein organischer Rest gebunden.[10] Verbindungen in denen an beiden Seiten der Carbonylgruppe ein organischer Rest gebunden ist (kein Wasserstoffatom) sind Ketone.[11] Eine Formylgruppe kommt außer in Aldehyden auch in der Ameisensäure und ihren Derivaten vor, die eine Carbonylgruppe mit einem Wasserstoffatom und einem Heteroatom (beispielsweise Sauerstoff) enthalten. Daher weist Ameisensäure einige Eigenschaften auf, die eigentlich für Aldehyde typisch sind. Beispielsweise wirkt sie im Gegensatz zu anderen Carbonsäuren reduzierend.[12] Verbindungen, die einen organischen Rest und ein Heteroatom an der Carbonylgruppe tragen werden allgemein als Carbonsäuren und Carbonsäurederivate (Ester, Amide und so weiter) bezeichnet.[13] Trägt die Carbonylgruppe zwei Heteroatome, handelt es sich um Kohlensäure beziehungsweise deren Derivate (Phosgen, Carbonate, Harnstoff und so weiter).[14]
Vertreter und Eigenschaften
[Bearbeiten | Quelltext bearbeiten]Die C=O-Doppelbindung in Aldehyden ist kurz und stark, beispielsweise bei Acetaldehyd mit einer Länge von 120,4 pm und einer Bindungsenergie von etwa 732–753 kJ/mol. Die Bindung ist außerdem polarisiert, Kohlenstoff- und Sauerstoffatom tragen Partialladungen.[6] Zwischen den Aldehydgruppen von Alkanalen kommt es zu Dipol-Dipol-Kräften, da die C=O-Doppelbindung sehr polar ist. Wasserstoffbrückenbindungen bilden sich nicht, weil kein sauerstoffgebundenes Wasserstoffatom vorhanden ist. Deswegen liegen die Siedepunkte der Aldehyde zwischen denen der Alkohole und Alkane. Mit Wasser können Aldehyde Hydrate bilden und Wasserstoffbrückenbindungen eingehen, weil das Sauerstoffatom zwei freie Elektronenpaare hat und negativ polarisiert ist. Deswegen sind kurzkettige Aldehyde gut wasserlöslich. Bei längerkettigen Aldehyden mit sechs oder mehr Kohlenstoffatomen überwiegt die Wirkung der unpolaren Alkylreste, was die Verbindungen praktisch unlöslich in Wasser macht. Viele Aldehyde haben einen charakteristischen und intensiven Geruch. So hat zum Beispiel Acrolein einen stechenden Geruch (wahrnehmbar beim Anbrennen von Fetten), wogegen Vanillin, Anisaldehyd und Zimtaldehyd angenehm riechen.[15]
Formaldehyd und Acetaldehyd sind bei Standardbedingungen gasförmig, Propnanal (C3) bis Hendecanal sind flüssig. Kurzkettige Aldehyde bis einschließlich Heptanal haben einen unengenehm stechenden Geruch, längerkettige von Octanal bis Tetradecanal riechen angenehm, noch längere sind geruchlos. Ungesättigte Aldehyde wie Acrolein oder Crotonaldehyd riechen im Vergleich zu den gesättigten Analoga noch unangenehmer, aromatische Aldehyde riechen im Allgemeinen angenehm.[16.1]
Spektroskopische Eigenschaften
[Bearbeiten | Quelltext bearbeiten]In IR-Spektren von Aldehyden und Ketonen findet man die intensive charakteristische Bande der C=O-Valenzschwingung im Bereich von 1690–1750 cm−1. Gesättigte aliphatische Aldehyde haben diese Bande bei 1740–1720 cm−1, Arylaldehyde bei 1715–1585 cm−1 und andere Aldehyde im Bereich von etwa 1790–1625 cm−1. Daneben liefern Aldehyde auch weniger intensive Bänder, zum Beispiel im Bereich 2900–2800 cm−1.[17]
In 13C-NMR-Spektren findet man das Signal des Carbonylkohlenstoffatoms von Aldehyden und Ketonen in einem Bereich von 195 und 210 ppm. Das dazugehörige Proton der Aldehydgruppe ist in 1H-NMR-Spektren als scharfes Signal bei etwa 10 ppm zu finden. Diese Eigenschaft macht die Identifikation mittels NMR-Spektroskopie besonders einfach, da in diesem hohen Bereich nur wenige Protonen eine Resonanz aufweisen.[6]
Vertreter
[Bearbeiten | Quelltext bearbeiten]Die Alkanale, also gesättigte, lineare Aldehyde, bilden eine homologe Reihe. Die allgemeine Summenformel der lautet CnH2nO (n = 1, 2, 3, 4, …). Die ersten vierzehn Vertreter sind in der folgenden Tabelle aufgelistet.
| Anzahl (C-Atome) |
IUPAC- Bezeichnung |
Trivialnamen | Summenformel | Strukturformel | Siedepunkt in °C[18] |
|---|---|---|---|---|---|
| 1 | Methanal | Formaldehyd | CH2O | 👁 Image |
0−19,1 |
| 2 | Ethanal | Acetaldehyd | C2H4O | 👁 Image |
−020,1 |
| 3 | Propanal | Propionaldehyd Propylaldehyd |
C3H6O | 👁 Image |
−048 |
| 4 | Butanal | n-Butyraldehyd | C4H8O | 👁 Image |
−074,8 |
| 5 | Pentanal | Valeraldehyd Amylaldehyd n-Pentaldehyd |
C5H10O | 👁 Image |
−103 |
| 6 | Hexanal | Capronaldehyd n-Hexaldehyd |
C6H12O | 👁 Image |
−131 |
| 7 | Heptanal | Önanthaldehyd Heptylaldehyd n-Heptaldehyd |
C7H14O | 👁 Image |
−152,8 |
| 8 | Octanal | Caprylaldehyd n-Octylaldehyd |
C8H16O | 👁 Image |
−171 |
| 9 | Nonanal | Pelargonaldehyd n-Nonylaldehyd |
C9H18O | 👁 Image |
−191 |
| 10 | Decanal | Caprinaldehyd n-Decylaldehyd |
C10H20O | 👁 Image |
−208,5 |
| 11 | Undecanal | Hendecanal n-Undecylaldehyd |
C11H22O | 👁 Image |
−117(18 mbar) |
| 12 | Dodecanal | Laurinaldehyd Dodecylaldehyd |
C12H24O | 👁 Image |
−238 |
| 14 | Tetradecanal | Myristylaldehyd Tetradecylaldehyd |
C14H28O | 👁 Image |
−260 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Neben den Alkanalen gibt es auch viele weitere Gruppen von Aldehyden, für die meistens historische Namen benutzt werden:
- Acrolein leitet sich von Propen – einem Alken – ab.
- Benzaldehyd leitet sich vom Benzol ab, ist also ein Arylaldehyd.
- Furfural (Furfurol, Furan-2-carbaldehyd) leitet sich von Furan ab, ist also ein Heteroarylaldehyd.[19]
Toxikologie
[Bearbeiten | Quelltext bearbeiten]Acetaldehyd ist kanzerogen und verursacht Mund-, Speiseröhren- und Magenkrebs. Unter anderem ist er bezogen auf die Menge ein wichtiger Inhaltsstoff in Tabakrauch und das wichtigste Karzinogen. Es spielt außerdem eine wichtige Rolle für die negativen gesundheitlichen Auswirkungen von Alkoholkonsum. Ethanol wird durch verschiedene Mikroorganismen durch Alkoholdehydrogenasen zu Acetaldehyd oxidiert. Viele Mikroorganismen sind aber weniger effizient im Abbau von Acetaldehyd, weshalb sich dieser anreichern kann.[20]
Aldehyde in der Luft verursachen vor allem Reizungen von Augen, Haut und Atemwegen. Die kurzkettigen wasserlöslichen reizen verstärkt Augen und obere Atemwege, längerkettige gelangen tiefer in die Atemwege und wirken auch auf die Lunge ein. Im Allgemeinen sind halogenierte und ungesättigte Aldehyde stärker reizend als gesättigte und diese wiederum stärker reizend als aromatische. Ähnliche Trends treten auch bei der Toxizität auf. Giftigkeit und Reizwirkung nehmen im Allgemeinen mit größerer Kettenlänge ab. Aldehyde wirken auch narkotisch, allerdings werden geringe Mengen schnell metabolisiert während bei höherer Dosis die Reizwirkung überwiegt. Aldehyde in Lösung können eine Sensibilisierung verursachen.[16.2] Formaldehyd verursacht beim Einatmen unter anderem Husten, Atembeschwerden und Kopfweh, bei hohen Konzentrationen auch Kehlkopf- oder Lungenentzündung. Bei wiederholter Exposition können chronische Reizungen der Atemwege und Augen auftreten, Hautkontakt mit Lösung oder Formaldehyd freisetzenden Produkten führen zu Sensibilisierung und Dermatitis.[16.3] Aldehyde werden für die durch Smog verursachten Augenreizungen mit verantwortlich gemacht, Formaldehyd ist neben Peroxyacetylnitrat eine Schlüsselverbindung hierfür.[16.4]
Acrolein wirkt stark reizend auf Augen und Atemwege; die Exposition führt unter anderem zu Tränenreizung, Atembeschwerden, Schläfrigkeit und Entzüngen von Rachen und Kehlkopf. Bei höheren Konzentrationen kann ein Lungenödem auftreten. Wiederholter Hautkontakt kann zu chronischen Reizungen, Dermatitis und in geringem Umfang zu Sensibilisierung führen.[16.5]
Vorkommen
[Bearbeiten | Quelltext bearbeiten]Terpenoide
[Bearbeiten | Quelltext bearbeiten]Ätherische Öle
[Bearbeiten | Quelltext bearbeiten]Terpenoide Aldehyde sind häufige Bestandteile von ätherischen Ölen. Geranial kommt beispielsweise in Geraniumöl, Zitronenöl, Zitronengrasöl sowie in ätherischen Ölen von Limette und Ingwer vor. Neral beispielsweise im Neroliöl, Rosenöl, Zitronenöl, Zitronengrasöl sowie in Ölen aus Zitronenmelisse, Ingwer und Bergamotte. Citronellal kommt in Kiefernöl und in Ölen aus Zitronenmelisse, Grapefruit, Zitrone, Ztitroneneucalyptus und Citronella (Cymbopogon nardus) vor.[21][21.1] Zitronengrasöl besteht überwiegend aus Aldehyden.[21.2] Myrtenal kommt in der Römischen Kamille vor.[21.3] Die Hauptaromakomponente von Safran ist das Safranal, ein Monoterpenaldehyd. Im frischen Zustand enthält die Pflanze stattdessen das bitter schmeckende Glycosid Picrocrocin, aus dem Safranal freigesetzt wird.[22] Perillaaldehyd ist Hauptbestandteil des Perillaöls aus der Perillapflanze.[23] Phellandral ist Bestandteil des Öls aus Wasserfenchel.[24][24.1] α-Sinensal und β-Sinensal kommen vor allem im Orangenöl vor.[24.2] Santalal ist eine wichtige Geruchskomponente im Sandelholzöl.[24.3]
Carotinoide
[Bearbeiten | Quelltext bearbeiten]Retinal ist eine essentielle Verbindung in der Lichtwahrnehmung von Tieren und Mikroorganismen, inklusive der Sehfähigkeit des Menschen. Die Lichtwahrnehmung basiert auf Proteinen, sogenannten Rhodopsinen, an die das Retinal in Form eines Imins gebunden ist. Das gebundene Retinal kann ein Photon aufnehmen, die dabei ausgelöste (E)/(Z)-Isomerisierung setzt dann eine Signalkaskade in Gang, die die Reizverarbeitung ermöglicht.[25] Neben Vitamin-A-Aldehyd (Retinal) kommen weitere aldehydische Carotinoide natürlich vor. Dazu gehören das in der Natur weit verbreitete β-Apo-8'-carotinal und andere längerkettige analoge Aldehyde als wahrscheinliche Vorläufer des Retinals aber auch Citraurin und Azafrinaldehyd.[26]
Aldehyde treten auch als Intermediate in der Biosynthese des Pflanzenhormons Abscisinsäure auf. 9-cis-Violaxanthin oder 9'-cis-Neoxanthin wird zunächst zum Aldehyd Xanthoxin gespalten. Dieser wird über Abscisinaldehyd in Abscisinsäure umgewandelt.[27]
Steroide und Saponine
[Bearbeiten | Quelltext bearbeiten]Aldosteron ist ein Wirbeltierhormon mit Aldehydfunktion, das unter anderem das Gleichgewicht diverser Elektrolyte im Blut reguliert. Es liegt unter gewöhnlichen Bedingungen zumeist als Halbacetal vor.[28] Einige Herzglycoside weisen eine Aldehydgruppe am C10 des Steroidgerüsts auf. Bei den typischen Glycosiden aus Digitalis steht dort eine einfache Methylgruppe, bei anderen Vertretern ist sie oft zu einer Hydroxymethyl- oder Formylgruppe oxidiert. Ein Beispiel für Verbindungen mit Formylgruppe sind die Glycoside des Strophanthidins und verwandter Aglycone. Dazu gehören unter anderem das Convallatoxin aus Maiglöckchen, Antiarin A und Antiarin B aus dem Upasbaum (Antiaris) und Cheirotoxin aus Goldlack. Das Frühlings-Adonisröschen enthält Adonitoxin und Cymarin; letzteres kommt auch in anderen Pflanzen beispielsweise der Gattung Hundsgift (Apocynum) vor.[29] Auch Saponine mit Aldehydfunktion sind bekannt, beispielsweise aus Quinoa, wobei sie im Vergleich zu Vertretern ohne Aldehydfunktion eine höhere Cytotoxizität aufweisen.[30]
Secologanin, Secoiridoide und Iridoide
[Bearbeiten | Quelltext bearbeiten]Secologanin ist ein Terpenoid, das eine Schlüsselverbindung in der Biosynthese vieler Alkaloid-Klassen mit insgesamt über 1000 Einzelverbindungen ist.[31][31.1] Dazu gehören Yohimbin und andere Indolalkaloide, Chinin und andere China-Alkaloide sowie Emetin und andere Ipecuanha-Alkaloide, aber auch die Secoiridoide wie das Sarracenin.[31.2] Oleocanthal ist ein in Olivenöl auftretendes Secoiridoid mit entzündungshemmenden Eigenschaften.[32]
Das Iridodial als Grundkörper der Iridoide entsteht aus Nerol oder Geraniol bei dem beide Kettenenden zum Aldehyd oxidiert werden, was 8-Oxogeranial ergibt, dessen Cyclisierung Iridodial ergibt. Weiteroxidation des Dialdehyds und Cyclisierung führt stattdessen zum Iridotrial, das wiederum weiter in Loganin und Secologanin umgewandelt wird.[31.3] Iridoide und Secoiridoide kommen hauptsächlich in Pflanzen vor, einige Vertreter wie das Iridodial kommen jedoch im Wehrsekret von bestimmten Ameisen vor.[31.2] Chrysomelidial ist eine verbreitete Verbindung bei Chrysomelinae (einer Unterfamilie der Blattkäfer) und bei Hornmilben (Oribatida). Zuerst isoliert wurde es aus dem Breiten Weidenblattkäfer (Plagiodera versicolora).[33] Weiterhin kommen in Wehrsekreten von Blattkäfern auch das strukturell verwandte Plagiodial sowie Salicylaldehyd vor.[34]
Sonstige Terpenoide
[Bearbeiten | Quelltext bearbeiten]Gossypol ist eine atropisomere Verbindung aus Pflanzen der Gattung Baumwolle (Gossypium) mit zwei Formylgruppen.[35] Callicarpenal ist ein als Repellent gegen Insekten wirkendes Terpenoid aus der Amerikanischen Schönfrucht. Die Aldehydfunktion der Verbindung ist dabei für die Wirkung nicht maßgeblich, der analoge Alkohol weist eine ähnliche Wirksamkeit auf.[36] In Schwertlilien kommen einige aldehydische Triterpenoide vor, beispielsweise Iridal und Irigermanal in der Deutschen Schwertlilie.[24.4]
Gyrinal ist ein Abwehrstoff aus Gyrinus natator (Gattung Taumelkäfer), das stark toxisch auf Mikroorganismen und Fische wirkt.[37] Botrytis cinerea, ein pflanzenpathogener Pilz, der die Grauschimmelfäule verursacht, bildet das phytotoxische Botrydial, einen reaktiven, terpenoiden Dialdehyd.[38]
Beim Wolligen Milchling (Lactarius vellereus) dient Stearoylvelutinal als Abwehrstoff. Bei Verletzung wird daraus das instabile Velutinal freigesetzt, die sich spontan in Velleral und Isovelleral umwandeln. Diese sind hochreaktive α,β-ungesättigte Aldehyde, die mit vielen Nucleophilen reagieren, beispielsweise Aminogruppen von Lysin in Enzymen, was vermutlich für die antibiotischen und cytotoxischen Eigenschaften verantwortlich ist. Ein analoger Verteidigungsmechanismus ist auch aus der Alge Caulerpa taxifolia bekannt, die den Ester Caulerpenin enthält, der den reaktiven Aldehyd Oxytoxin freisetzt. Auch andere Pilze enthalten terpenoide Ester, die bei Verletzung des Fruchtkörpers Aldehyde freisetzen, so der Lärchen-Reizker (Lactarius porninsis), der den Aldehyd Porninsal bildet, sowie der Edel-Reizker (Lactarius deliciosus) und der Fichten-Reizker (Lactarius deterrimus), die Lactaroviolin bilden. Bei diesen Pilzen ist aber nicht klar, inwieweit es sich um einen Verteidigungsmechanismus handelt, insbesondere, da die beiden Lactaroviolin-bildenden Arten ungiftige Speisepilze sind.[39]
Kohlenhydrate
[Bearbeiten | Quelltext bearbeiten]Monosaccharide sind polyhydroxylierte Aldehyde (Aldosen) und Ketone (Ketosen); Oligo- und Polysaccharide sind abgeleitete Oligomere und Polymere, wobei die Carbonylgruppen in Acetale umgewandelt sind. Sehr weit verbreitete Aldosen sind Glucose, Galactose und Mannose. Ribose und Desoxyribose sind Bausteine von RNA und DNA. Andere Vertreter wie Arabinose, Xylose und Rhamnose sind Bausteine von Pflanzengummi, Hemicellulosen und Glycosiden. In ungebundener Form liegen die Aldosen im Gleichgewicht von Aldehyd und cyclischem Halbacetal vor. Die kristallinen Feststoffe liegen meist als Halbacetale vor; auch im Gleichgewicht in Lösung herrscht diese Form vor.[40][40.1] Streptose ist eine Aldose mit verzweigter Kohlenstoffkette und weist eine zweite Formylgruppe auf. Sie kommt als Baustein des Glycosids Streptomycin vor.[41]
Aldehyde treten als Intermediate in Fermentationsprozessen durch Hefen und Bakterien auf. Bei der alkoholischen Gärung durch Saccharomyces cerevisiae wird zunächst Glucose in zwei Einheiten Pyruvat gespalten. Anschließend wird Pyruvat zu Acetaldehyd decarboxyliert und dieser zu Ethanol reduziert.[42][42.1] Enterobacteriaceae und Clostridrien bilden Ethanol über die Reduktion von Acetyl-CoA zu Acetaldehyd und dann zu Ethanol.[42.2] Einige Milchsäurebakterien bilden Ethanol über den Pentosephosphatweg. Dabei wird zunächst Xylulose-5-phosphat gebildet und dieses in Glyceraldehyd-3-phosphat und Acetylphosphat gespalten; letzteres wird über Acetyl-CoA und Acetaldehyd zu Ethanol reduziert.[42.3]
Aminosäuren und Derivate
[Bearbeiten | Quelltext bearbeiten]Aminosäuren mit Aldehydfunktion
[Bearbeiten | Quelltext bearbeiten]Sulfatasen vom Typ I, die Schwefelsäureester hydrolysieren, sind über alle Lebewesen hochkonserviert und enthalten die Aminosäure Formylglycin. Diese entsteht als posttranslationale Modifikation durch Oxidation von Cystein oder Serin. Der Mechanismus der katalysierten Reaktion basiert vermutlich auf der Bildung eines Aldehydhydrats, in dem eine Hydroxygruppe als Nucleophil wirkt, das einen Schwefelsäureester angreifen kann. Dabei entsteht ein Ester des hydratisierten Formylglycins mit Schwefelsäure; durch Eliminierung von Hydrogensulfat wird die Formylgruppe zurückgebildet und kann wieder hydratisiert werden.[43] Aldehyde spielen weiterhin eine Rolle bei der Quervernetzung von Elastin und Kollagen. Dabei werden Lysin und Hydroxylysin durch Lysin-6-oxidase in die entsprechenden Aldehyde umgewandelt, also Allysin aus Lysin und Hydroxyallysin aus Hydroxylysin. Ersteres tritt vor allem in der Haut auf, letzteres in Knochen und Knorpeln. Die Aldehydgruppen ermöglichen dann die Ausbildung kovalenter Bindungen zwischen Proteinsträngen, unter anderem durch Aldolreaktionen. Im Falle von Elastin wird unter anderem aus Allysin das Desmosin und das Isodesmosin als Quervernetzter gebildet.[44]
Alkaloide
[Bearbeiten | Quelltext bearbeiten]4-Hydroxyphenylacetaldehyd, das aus Tyrosin gebildet wird, ist ein Intermediat in der Biosynthese von Morphin und anderen Benzylisochinolin-Alkaloiden.[45] Auch die Biosynthese anderer Alkaloide aus Aminosäuren verläuft über Aldehyde. Die Chinolizidin-Alkaloide wie das Spartein entstehen aus Lysin; dieses wird zunächst zu Cadaverin decarboxyliert und dann in 5-Aminopentanal umgewandelt. Drei Moleküle dieser Verbindung ergeben ein tetracyclisches Grundgerüst. Pyrrolizidinalkaloide entstehen ausgehend von Ornithin beziehungsweise Arginin über Homospermidin. Die Umwandlung des Homospermidins in die bicyclische Necinbase als Grundgerüst der Alkaloide verläuft wiederum über einen Aldehyd.[46] Hydroxydanaidal dient bei Schmetterlingen der Art Utetheisa ornatrix sowohl als Pheromon als auch als Abwehrstoff: Es ist ein männliches Sexualpheromon; zusätzlich wird es bei der Paarung ans Weibchen übertragen; Hydroxydanaidal von beiden Elternteilen gelangt in die Eier und dient als Fraßschutz. Gebildet wird es aus Pyrrolizidinalkaloiden wie Monocrotalin-N-oxid oder Heliotrin, die mit der Nahrung aufgenommen werden.[47]
Peptide
[Bearbeiten | Quelltext bearbeiten]Peptidische Aldehyde wirken als Inhibitoren von Proteasen, beispielsweise das aus Streptomyces isolierte α-MAPI. Die Aldehydfunktion ist dabei essentiell für die inhibitorische Wirkung, Reduktion zum Alkohol oder Oxidation zur Carbonsäure führt zum Wirkungsverlust.[48] Verwandte Peptide mit ähnlicher Wirkung sind Antipain und Leupeptin, die Arginal enthalten, den vom Arginin abgeleiteten Aldehyd.[49]
Sonstige
[Bearbeiten | Quelltext bearbeiten]Aldehyde kommen als Abbauprodukte cyanogener Glycoside vor. Bei deren Hydrolyse entsteht zunächst ein Cyanhydrin, das spontan Cyanwasserstoff freisetzt, wobei gleichzeitig je nach Struktur der Verbindung ein Aldehyd oder Keton entsteht. Benzaldehyd ist die Carbonylkomponente aus Amygdalin, Prunasin und Vicianin. 4-Hydroxybenzaldehyd ist die Carbonylkomponente von Dhurrin und Taxiphyllin.[50] Die Biosynthese der cyanogenen Glycoside verläuft jedoch nicht über Aldehyde, sondern über Aldoxime, die aus Aminosäuren gebildet und dann entweder direkt oder über ein Nitril in ein Cyanhydrin überführt werden.[51]
Derivate von Phenylalanin sind eine der wichtigsten Klassen flüchtiger Verbindungen aus Pflanzen. Phenylacetaldehyd wird durch die bifunktionelle Phenylacetaldehyd-Synthase direkt aus Phenylalanin gebildet.[52]
Fettaldehyde und Abbauprodukte von Fettsäuren
[Bearbeiten | Quelltext bearbeiten]Semiochemikalien
[Bearbeiten | Quelltext bearbeiten]Die Sexualphermone diverser Insekten aus den Ordnungen Käfer (Coleoptera) und Schmetterlinge (Lepidoptera) sind ungesättigte Verbindungen, wobei aliphatische Kohlenwasserstoffe, Epoxide, Alkohole, Ketone, Carbonsäureester und auch Aldehyde auftreten.[53] Bei Motten sind die Pheromone häufig von Fettsäuren abgeleitete Fettalkohole, Acetate und Fettaldehyde mit 10–18 Kohlenstoffatomen.[54][54.1] Dabei wird zunächst ein Acyl-CoA-Verbindung mit bestimmter Kettenlänge gebildet und gegebenenfalls eine oder mehrere Doppelbindungen eingeführt, dann die Verbindung enzymatisch zu einem Alkohol reduziert.[54.2] Aldehyde entstehen durch enzymatische Reoxidation dieser Alkohole.[54.3] Beispielsweise enthalten die Sexualpheromone sowohl von Chrysoteuchia topiaria als auch von Diatraea grandiosella (beide Familie Rüsselzynsler) als Hauptkomponente (Z)-11-Hexadecenal sowie als Nebenkomponente (Z)-9-Hexadecenal.[53] Weitere Beispiele für aldehydische Bestandteile von Sexualpheromonen sind Nonanal und Undecanal bei der Großen Wachsmotte,[55] Phytanal bei Eurema mandarina (Familie Weißlinge),[56] Hexadecanal bei Bicyclus anynana (Familie Edelfalter).[56] Bei den Hymenoptera treten zum Teil reine Kohlenwasserstoffe als Pheromone auf, wobei Aldehyde als biosynthetisches Intermediat vorkommen. Diese werden gebildet über ein Acyl-CoA, Reduktion zum Fettalkohol, Oxidation zum Aldehyd und Decarbonylierung zum Kohlenwasserstoff.[54.4] Hexadecanal wurde neben dem Vorkommen in Insekten auch als Semiochemikalie bei Menschen identifiziert, die das Aggressionsverhalten beeinflusst.[57]
Lipoxygenase-Weg und Lipidperoxidation
[Bearbeiten | Quelltext bearbeiten]Ein wichtiger Prozess, mit dem Pflanzen auf widrige Umgebungsbedingungen reagieren, ist der Lipoxygenase-Pathway. Dabei werden ungesättigte Fettsäuren, insbesondere Linolsäure und α-Linolensäure durch eine Lipoxygenase in Position 9 oder 13 zu einem Hydroperoxid oxidiert, wobei mehr Enzyme bekannt sind, die 13-Hydroperoxide bilden. Diese Hydroperoxide reagieren dann weiter zu diversen oxygenierten Verbindungen, sogenannten Oxylipinen. Dazu gehören beispielsweise Jasmonate oder oxygenierte Fettsäuren. Oxylipine erfüllen verschiedene biologische Funktionen, unter anderem zur Abwehr von Pathogenen. Durch Hydroperoxid-Lyasen werden die Hydroperoxide unter anderem zu Aldehyden und Alkoholen umgesetzt.[58] Zu den aus den 13-Hydroperoxiden gebildeten Verbindungen gehören die grünen Blattduftstoffe. Dabei handelt es sich um C6-Aldehyde, -alkohole sowie deren Ester, die für den typischen Geruch beispielsweise beim Schneiden von Blättern verantwortlich sind. Oxidation von Linolensäure ergibt beispielsweise 13-Hydroperoxy-9,11,15-octadecatriensäure, dessen Spaltung durch eine Hydroperoxid-Lyase (Z)-3-Hexenal ergibt, das sich leicht zu (E)-2-Hexenal umlagert. Ausgehend von Linolsäure wird Hexanal gebildet. Die 9-Hydroperoxide der Fettsäuren werden zu je zwei C9-Verbindungen gespalten, wobei neben anderen Oxidationsprodukten ebenfalls Aldehyde anfallen. Die meisten der auf diesem Weg gebildeten Oxylipine sind stark bakterizid, insbesondere (E)-3-Hexenal und (Z)-3-Hexenal.[59] Die im Lipoxygenase-Weg gebildeten C6- und C9-Aldehyde sind auch Aromakomponenten diverser Pflanzen, beispielsweise in Äpfeln.[60] (Z)-3-Hexenal und (E)-2-Hexenal sind wichtige Aromastoffe in Kirschtomaten und (E, Z)-2,6-Nonadienal ist die Hauptaromakomponente in Gurken.[61] Traumatin, das ebenfalls im Lipoxygenaseweg gebildet wird, ist ein Pflanzenhormon, das insbesondere bei Verletzungen freigesetzt wird und die Zellteilung anregt.[62] Bei Kieselalgen spielen die Oxylipine, inklusive mehrfach ungesättigter Aldehyde, eine Rolle in der Verteidigung gegen Ruderfußkrebse als Fressfeinde.[63]
Auch bei der Lipidperoxidation werden aus Fettsäuren über Peroxide Aldehyde gebildet. Dazu gehört das für den charakteristischen Geruch von Blut verantwortliche trans-4,5-epoxy-(E)-2-decenal, das Raubtiere anlockt, während sich mögliche Beutetiere dadurch entfernen beziehungsweise in erhöhte Alarmbereitschaft versetzt werden.[64]
Glyoxylatzyklus
[Bearbeiten | Quelltext bearbeiten]Der Glyoxylatzyklus ist ein Stoffwechselweg der sich teilweise mit den Citratzyklus überschneidet und über Glyoxylat verläuft. Statt decarboxyliert zu werden, wird dabei Isocitrat durch die Isocitrat-Lyase in Succinat und Glyoxylat gespalten. Letzteres wird durch Reaktion mit Acetyl-CoA in Malat umgewandelt. Im Gesamtzumsatz werden zwei Einheiten Acetyl-CoA in ein Oxalacetat überführt, was die Umwandlung von Fettsäuren in Kohlenhydraten ermöglicht. Der Glyoxylatzyklus kommt vor allem bei Bakterien, Hefen und Pflanzen vor.[65]
Biolumineszenz bei Bakterien
[Bearbeiten | Quelltext bearbeiten]Biolumineszente Bakterien (Gattungen Vibrio, Photobacterium und Xenorhabdus) nutzen Fettaldehyde als Substrat für die Lichterzeugung. Dabei werden unter Einwirkung einer Luciferase reduziertes Flavinmononukleotid und ein Aldehyd mit molekularem Sauerstoff oxidiert, wobei grünblaues Licht emittiert wird. Verschiedene Untersuchungsergebnisse deuten darauf hin, dass natürlicherweise vor allem Tetradecanal umgesetzt wird. Andererseits gibt es Unterschiede zwischen verschiedenen Bakterienarten und deren Luciferasen und einige darunter erzielen sehr hohe Lichtausbeuten mit Nonanal, Decanal oder Dodecanal. Die Bakterien verfügen über einen Enzymkomplex, der die Reduktion von Fettsäure zu Aldehyd katalysiert.[66]
Sonstige biogene Aldehyde
[Bearbeiten | Quelltext bearbeiten]Pyridoxalphosphat (Vitamin B6) ist ein Cofaktor, der insbesondere eine wichtige Rolle im Metabolismus von Aminosäuren spielt. Bei diesen Reaktionen, zu denen Transaminierungen, Racemisierungen und Decarboxylierungen gehören, wird in der Regel zunächst aus einer Pyridoxalverbindung und einer Aminosäure ein Imin gebildet.[67]
Zimtaldehyd ist eine charakteristische Aromaverbindung von Zimt und Zimtkassie, wobei letztere deutlich mehr enthält.[21.1] Das Öl der Zimtkassie gehört zu den ätherischen Ölen, die hauptsächlich aus Aldehyden bestehen.[21.2] Weitere aromatische Aldehyde sind Anisaldehyd aus Anis und Cuminaldehyd aus Kreuzkümmel.[21.3] Piperonal kommt als Aromaverbindung in schwarzem Pfeffer vor. Gebildet wird es aus Ferulasäure, indem zunächst die Benzodioxoleinheit aufgebaut und dann die Aldehydgruppe durch Spaltung der C=C-Doppelbindung erzeugt wird.[68] Vanillin, die charakteristische Aromakomponente der Vanille, ist ein Aldehyd.[21.3] Vanillin wird vermutlich ebenfalls durch Doppelbindungsspaltung aus Ferulasäure biosynthetisiert. In den grünen Schoten liegt es in Form des Vanillinglucosids ohne den charakteristischen Geschmack vor.[69] Alkanale kommen ebenfalls in ätherischen Ölen vor, jedoch zu geringen Anteilen von meist unter 1 %. Dazu gehören Octanal (Zitrone, Mandarine, Rose), Nonanal (Zitrone, Orange, Grapefruit), Decanal (Zitronengras, Mandarine, Petitgrain) und Undecanal (Zitrone, Bitterorange).[21.4]
Atranorin und Fumarprotocetrarsäure sind aromatische Aldehyde aus Flechten.[70]
Die Aldehyde (E, Z)-2,6-Nonadienal und (E)-2-Nonenal kommen als Pheromone bei der Deutschen Skorpionsfliege vor.[71] Bei Bettwanzen wirken (E)-2-Hexenal und (E)-2-Octenal sowohl zur Abwehr als auch als Pheromon.[72]
Josamycin und verwandte Verbindungen sind antibakteriell und cytotoxisch wirksame Makrolid-Antibiotika, die eine Aldehydgruppe enthalten, die aber für die biologische Wirkung nicht essentiell ist.[73] Ein anderes antibakterielles Makrolid mit Aldehydgruppe ist das Tylosin, von dem durch Modifikation der Aldehydgruppe Derivate erhalten wurden, die verbesserte antibiotische Wirksamkeit aufweisen.[74] Formaldehyd ist ein zentrales Intermediat im Stoffwechsel methylotropher Mikroorganismen. Diese Organismen können reduzierte C1-Verbindungen als einzige Energie- und Kohlenstoffquelle nutzen. Dabei werden beispielsweise Methan, Methanol, Dichlormethan oder Methansulfonsäure zunächst in Formaldehyd umgewandelt, welcher als Substrat im Ribulosemonophosphatweg und anderen Stoffwechselwegen dient.[75]
Nichtbiogene Vorkommen
[Bearbeiten | Quelltext bearbeiten]Abbau von Kohlenhydraten und Aminosäuren
[Bearbeiten | Quelltext bearbeiten]Das Erhitzen von Monosacchariden unter sauren Bedingungen führt zur Bildung von dehydratisierten Produkten wie Furanen. In einigen Fällen bleibt dabei eine Aldehydfunktion einer Aldose erhalten, beispielsweise bei der Bildung von Furfural, 5-Hydroxymethylfurfural und 5-Methylfurfural.[40.2] Beim Erhitzen von Aldosen in Gegenwart von Aminen, insbesondere Aminosäuren, findet die Maillard-Reaktion unter Bildung von braunen Farbstoffen (Melanoidinen) und Aromaverbindungen statt. Diese beginnt mit der Bildung eines Imins aus Aldose und Amin und führt über eine Amadori-Umlagerung zu einem Aminoketon. Dieses kann wiederum zu einer Dicarbonylverbindung abgebaut werden.[40.3] Die Dicarbonylverbindungen können mit Aminosäuren in einem Strecker-Abbau mit oxidativer Decarboxylierung Aldehyde bilden. Dies passiert vor allem beim Erhitzen von Lebensmitteln einem höheren Gehalt an freien Aminosäuren und spielt eine wichtige Rolle für das Aroma von Speisen. So gebildete Aldehyde sind vor allem Methional, Phenylacetaldehyd, 3-Methylbutanal, 2-Methylbutanal und Isobutanal.[40.4]
Atmosphärische Vorkommen
[Bearbeiten | Quelltext bearbeiten]Die wichtigste Quelle von Aldehyden in der Atmosphäre sind unvollständige Verbrennungsprozesse von Kohlenwasserstoffen, vor allem in Abgasen von Fahrzeugen. Daneben spielt aber auch die Freisetzung aus industriellen Prozessen eine Rolle sowie die Neubildung in der Atmosphäre durch Reaktion von Kohlenwasserstoffen und Stickoxiden. Von besonderer Bedeutung im Zusammenhang mit Luftverschmutzung sind Formaldehyd und Acrolein.[16][16.6] Atmosphärische Reaktionen, die Aldehyde bilden, treten beispielsweise zwischen Alkenen und Stickoxiden, Ozon oder Sauerstoff sowie zwischen Aromaten und Stickoxiden auf. Formaldehyd im Besonderen tritt als Produkt der Photooxidation fast aller Alkene und Aromaten auf sowie als Abbauprodukt höherer Aldehyde. Acrolein entsteht als Oxidationsprodukt von Alkenen mit mehreren Doppelbindungen.[16.7]
Extraterrestrische Vorkommen
[Bearbeiten | Quelltext bearbeiten]Formaldehyd wurde als erste organische Verbindung mit mehr als zwei Atomen im interstellaren Medium gefunden und die Entdeckung 1969 publiziert. Vorher waren lediglich zweiatomige Spezies und anorganische Verbindungen (Wasser, Ammoniak) bekannt.[76] Aldehyde sind in Sternentstehungsregionen und Meteoriten vom Typ der kohligen Chondriten weit verbreitet. Stand 2024 waren neun verschiedene Aldehyde aus dem interstellaren Medium bekannt, neben Formaldehyd auch dessen protonierte Form, außerdem Acetaldehyd, Propinal, Acrolein, Propanal, Formylcyanid, 3-Hydroxypropenal und Glycolaldhyd. Sechzehn Vertreter waren aus kohligen Chondriten bekannt: Neben Formaldehyd, Acetaldehyd und Propanal auch Isobutanal, Butanal, Pentanal, Isopentanal, 2-Methylbutanal, Pivalaldehyd, Hexanal, Isohexanal, 2-Methylpentanal, 3-Methylpentanal, 2-Ethylbutanal, 2,2-Dimethylbutanal und 3,3-Dimethylbutanal.[77]
Bedeutung für die Entstehung des Lebens
[Bearbeiten | Quelltext bearbeiten]Für Formaldehyd wird eine zentrale Rolle in der Entstehung biologisch relevanter Moleküle angenommen, da es einerseits leicht aus verschiedenen Bedingungen gebildet werden kann, andererseits auch leicht verschiedene komplexere Moleküle bildet, beispielsweise Glycin, durch Reaktion mit Glycin andere Aminosäuren, sowie unter basischen Bedingungen Zucker.[78] Der Aufbau von komplexeren Molekülen aus einfachen Vorläufern unter milden Reaktionsbedingungen ist unter thermodynamischen und kinetischen Gesichtspunkten nur mit Carbonylverbindungen möglich (beispielsweise durch Aldolreaktionen), da nicht wie bei heutigen Lebensformen Enzyme vorhanden waren, die Energie für thermodynamisch ungünstige Reaktionen umverteilen können oder auch Reaktionen ermöglichen, die kinetisch ansonsten unmöglich sind. Die einzige C-, O- und H-haltige C1-Verbindung für derartige Reaktionen ist demnach Formaldehyd.[79] Die Strecker-Synthese von Aminosäuren aus einem Aldehyd, Ammoniak und Cyanwasserstoff über ein Nitril wird allgemein als plausibler Mechanismus für die Entstehung dieser Verbindungsklasse angenommen. Andererseits ist die Formosereaktion ein möglicher Entstehungsweg für Kohlenhydrate, wobei Formaldehyd unter alkalischen Bedingungen oligomerisiert; im ersten Schritt wird Glycolaldehyd gebildet. Auch die Bildung von Glycerin als Baustein der Triglyceride ist aus Formaldehyd möglich.[80]
Herstellung
[Bearbeiten | Quelltext bearbeiten]Aldehyde können aus vielen verschiedenen Vorläufern mit diversen Methoden gewonnen werden. Oxidationen primärer Alkohole, mit Swern-Oxidation und Anelli-Oxidation als wichtige Beispiele, sind besonders vielfältig und finden breite Anwendung. Daneben spielen Oxidationen anderer Vorläufer, die Reduktion von Carbonsäurederivaten und Formylierungen, bei denen die gesamte Aldehydgruppe inklusive des Kohlenstoffatoms eingeführt wird, eine Rolle. Industriell wichtige Verfahren sind insbesondere die Hydroformylierung terminaler Alkene und die Wacker-Oxidation von Ethylen zu Acetaldehyd.
Oxidation primärer Alkohole
[Bearbeiten | Quelltext bearbeiten]In der organischen Synthese ist die Oxidation primärer Alkohole die häufigste Synthese für Aldehyde.[81] Weit verbreitete Oxidationsmethoden zur Herstellung von Aldehyden basieren auf Chrom(VI)-verbindungen, Mangan(IV)-oxid, aktivierten Derivaten von Dimethylsulfoxid, hypervalenten Iod-Verbindungen, Ruthenium-Verbindungen und Nitroxylradikalen.[82][83.1]
{kind=link}
{kind=link}
Chrom(VI) als Oxidationsmittel
[Bearbeiten | Quelltext bearbeiten]Verschiedene Chrom(VI)-Reagenzien eignen sich für die Oxidation von Alkoholen zu Aldehyden und Ketonen. Die selektive Oxidation von primären Alkoholen zu Aldehyden gelingt dabei nur unter wasserfreien Bedingungen. In Anwesenheit von Wasser bilden sich nach dem ersten Oxidationsschritt Aldehydhydrate, die zur Carbonsäure weiteroxidiert werden, während die Aldehyde selbst gegenüber den Chrom(VI)-Reagenzien stabil sind.[84.1] Eine geeignete Verbindung ist der Komplex aus einem Molekül Chrom(VI)-oxid mit zwei Molekülen Pyridin. Wird Pyridin auch als Lösungsmittel verwendet, wird die Reaktion als Sarett-Oxidation bezeichnet, mit Dichlormethan als Collins-Reaktion. Neuer ist die Ratcliffe-Variante, wobei der Komplex in situ erzeugt wird, indem Chrom(VI)-oxid unter Rühren zu einer Lösung von Pyridin in Dichlormethan gegeben wird. Dadurch wird das direkte Arbeiten mit dem Komplex vermieden, bei dem es leicht zu Explosionen kommen kann.[82.1] Weitere Chrom(VI)-Reagenzien sind Pyridiniumdichromat und Pyridiniumchlorochromat, die beide in wässriger Lösung hergestellt werden, dabei jedoch ausfallen und in getrockneter Form unter wasserfreien Bedingungen in Dichlormethan eingesetzt werden können.[82.2] Die Jones-Oxidation mit Chrom(VI)-oxid und Schwefelsäure ist wegen der Anwesenheit von Wasser oft nicht zur Herstellung von Aldehyden geeignet, kann jedoch zur Herstellung flüchtige Aldehyde verwendet werden, wenn das Produkt während der Reaktion abdestilliert wird.[82.3] Beispielsweise kann 1-Propanol so oxidiert werden und das entstehende Propanal abdestilliert werden.[83.2]
Aktiviertes Dimethylsulfoxid als Oxidationsmittel
[Bearbeiten | Quelltext bearbeiten]Bei der Pfitzner-Moffatt-Oxidation greift Dimethylsulfoxid (DMSO) über das Sauerstoffatom ein protoniertes Dicyclohexylcarbodiimid (DCC) an und bildet eine Sulfoniumverbindung. Bei einer anschließenden Substitution durch den Alkohol am Schwefel wird das DCC als Dicyclohexylharnstoff wieder abgespalten, wobei ein Schwefel-Ylid entsteht. Aus diesem bildet sich durch intramolekulare Eliminierung von Dimethylsulfid der Aldehyd.[82.4]
Eine Weiterentwicklung der Pfitzner-Moffatt-Oxidation mit sehr verbreiteter Anwendung ist die Swern-Oxidation, bei der statt DCC Oxalylchlorid verwendet wird. Nachteile sind hier die Bildung von hochgiftigem Kohlenmonoxid und dass die Reaktion zum Erzielen einer guten Selektivität nur bei niedrigen Temperaturen durchgeführt werden kann. Dafür werden mit der Swern-Oxidation meist sehr gute Ausbeuten erzielt. Andere Varianten sind die Albright-Goldman-Oxidation (mit Acetanhydrid statt DCC), die Parikh-Doering-Oxidation (mit Schwefeltrioxid-Pyridin-Komplex statt DCC), die Albright-Onodera-Oxidation (mit Phosphorpentoxid statt DCC), sowie eine Variante mit Trifluoressigsäureanhydrid statt DCC.[82.4] Viele weitere Elektrophile können DMSO aktivieren, dazu gehören beispielsweise Mesylchlorid, Phosphortrichlorid, Phosphorylchlorid, Triphenylphosphindichlorid, Triphenylphosphindibromid und Acetylbromid.[82.5]
{kind=link}
{kind=link}
Auch bei der Corey-Kim-Reaktion findet ein aktiviertes Derivat von DMSO Verwendung, allerdings wird dieses nicht aus DMSO selbst hergestellt, sondern durch Oxidation von Dimethylsulfid mit elementarem Chlor, wobei Chlordimethylsulfoniumchlorid entsteht (dieselbe Verbindung wie bei der Swern-Oxidation). Eine Variante, die die gefährliche Verwendung von gasförmigem Chlor vermeidet, ist die Oxidation von Dimethylsulfid mit N-Chlorsuccinimid (NCS), wobei das reaktive Intermediat statt einem Chloratom eine Succinimid-Gruppe trägt.[82.6]
Hypervalente Iod-Verbindungen als Oxidationsmittel
[Bearbeiten | Quelltext bearbeiten]{kind=link}
{kind=link}
Ein verbreitetes Oxidationsmittel zur Herstellung von Aldehyden sind hypervalente Iodverbindungen, bei denen ein Iodatom in einer hohen Oxidationsstufe vorliegt. Früher bekannte hypervalente Iodverbindungen waren sehr instabil und zudem meist kaum bis gar nicht in organischen Lösungsmittel löslich, was ihre Anwendung stark einschränkte. Das im Jahr 1983 präsentierte Dess-Martin-Periodinan ist eine Verbindung, die beide Probleme löst, weshalb sie oft für die Herstellung von Aldehyden verwendet wird. Die Reaktion wird oft in Dichlormethan durchgeführt. Als Nebenprodukt entsteht aus dem Oxidationsmittel auch Essigsäure, die aber mittels Pyridin oder Natriumhydrogencarbonat neutralisiert werden kann. Eine weitere hypervalente Iodverbindung, Iodoxybenzoesäure ist schon deutlich länger bekannt, ist aber in den meisten organischen Lösungsmitteln unlöslich. Geeignete Reaktionsbedingungen, insbesondere der Einsatz von Dimethylsulfoxid als Lösungsmittel, wurden erst nach der Entdeckung des Dess-Martin-Periodinans entwickelt.[82.7]
Nitroxylradikale als Oxidationsmittel
[Bearbeiten | Quelltext bearbeiten]{kind=link}
{kind=link}
Die Anelli-Oxidation von Alkoholen zu Aldehyden und Ketonen nutzt eine katalytische Menge eines Nitroxylradikals wie TEMPO als direktes Oxidationsmittel, Natriumhypochlorit als stöchiometrisches Oxidationsmittel, Kaliumbromid als Beschleuniger (vermutlich durch Bildung von hypobromiger Säure) und Natriumhydrogencarbonat zum Absenken des pH-Werts, in der Regel in einem zweiphasigen Lösungsmittelgemisch aus Wasser und Dichlormethan. Primäre Alkohole werden bei 0 °C innerhalb von Minuten zu Aldehyden oxidiert. Neben TEMPO sind auch andere Derivate wie 4-Methoxy-TEMPO und 4-Acetamido-TEMPO als Reagenzien geeignet. Auch andere stöchiometrische Oxidationsmittel als Natriumhypochlorit können verwendet werden.[82.8]
Andere Oxidationsmittel
[Bearbeiten | Quelltext bearbeiten]Perruthenat-Ionen oxidieren Wasser, während Natriumperruthenat und Kaliumperruthenat in organischen Lösungsmitteln unlöslich sind, weshalb vor allem das in organischen Lösungsmitteln lösliche Tetrapropylammoniumperruthenat (TPAP) als gängiges Reagenz für die Oxidation von Alkoholen zu Aldehyden und Ketonen dient (Ley-Oxidation). In der Regel wird eine katalytische Menge verwendet (zum Beispiel 5 mol-%) und N-Methylmorphin-N-oxid als stöchiometrisches Oxidationsmittel zugesetzt (zum Beispiel 1,5 Äquivalente).[82.9]
Bei der Fétizon-Oxidation wird Silbercarbonat auf Kieselgur (Fétizon-Reagenz) als Oxidationsmittel verwendet. Es handelt sich um einen Prozess der heterogenen Katalyse, bei dem der zu oxidierende Alkohol am Silbercarbonat adsorbiert wird. Andere polare funktionelle Gruppen und selbst mäßig polare Lösungsmittel wie Ethylacetat können bei der Adsorption mit der Alkoholgruppe konkurrieren, weshalb dei Reaktion in der Regel in kochendem Benzol durchgeführt wird.[82.10]
Mangan(IV)-oxid als Oxidationsmittel kann durch Umsetzung von Mangan(II)-sulfat mit Kaliumpermanganat und Natriumhydroxid gewonnen werden. Es ist ein selektives Oxidationsmittel für Hydroxygruppen in allylischer oder benzylischer Position. So können Hydroxymethylgruppen an Doppelbindungen und aromatischen Ringen zu Aldehyden oxidiert werden.[85.1]
Die Oppenauer-Oxidation, bei der ein Alkohol zu einer Carbonylverbindung oxidiert wird, während eine andere Carbonylverbindung als Oxidationsmittel dient, ist für die Herstellung von Aldehyden thermodynamisch ungünstig, da sie im Vergleich zu anderen Carbonylverbindungen höhere Oxidationspotentiale aufweisen.[82.11]
{kind=link}
{kind=link}
Oxidation anderer Edukte
[Bearbeiten | Quelltext bearbeiten]Alkylhalogenide
[Bearbeiten | Quelltext bearbeiten]Die Oxidation von Alkylhalogeniden zu Aldehyden und Ketonen mit Dimethylsulfoxid (DMSO) wird Kornblum-Oxidation genannt, aus primären Halogeniden sind Aldehyde zugänglich. Bei hochreaktiven Edukten, beispielsweise Benzylbromiden mit elektronenarmem Ring, findet die Reaktion schon beim Zugeben von DMSO bei Raumtemperatur statt. In den meisten Fällen ist jedoch die Herstellung eines Tosylats (mit Silbertosylat) und Erhitzen nötig. Die Oxidation mit Trimethylamin-N-oxid und DMSO (Ganem-Oxidation) funktioniert ohne Aktivierung bei Raumtemperatur.[86.1] Primäre Alkylhalogenide können außerdem mit Pyridin-N-oxid oder 2-Picolin-N-oxid zu Aldehyden oxidiert werden.[87.1] Bei der Hass-Bender-Oxidation dient 2-Nitropropan als Oxidationsmittel zur Oxidation von Benzylhalogeniden zu Aldehyden.[87.2]
In der Kröhnke-Reaktion wird ein Alkylhalogenid durch Umsetzung mit Pyridin substituiert und mit einer Base in ein Stickstoff-Ylid überführt. Oxidation mit N,N-Dimethyl-4-nitrosoanilin ergibt ein Nitron und dessen Hydrolyse ein Aldehyd. Die Methode ist recht limitiert, was die geeigneten Edukte angeht, sie eignet sich aber gut zur Herstellung enderweitig schwierig zugänglicher heterocyclischer Aldehyde.[87.3]
Bei der Sommelet-Reaktion (verwandt mit der Duff-Reaktion) werden primäre aromatische Halogenide (Benzylhalogenide) mit Urotropin zu Aldehyden umgesetzt.[88.1]
{kind=link}
{kind=link}
Methylgruppen
[Bearbeiten | Quelltext bearbeiten]Methylgruppen in α-Stellung zu einer Carbonylgruppe können mittels Selendioxid zur Formylgruppe (Aldehyd) oxidiert werden (Riley-Oxidation).[89.1] Bei der Étard-Reaktion wird eine an einen Aromaten gebundene Methylgruppe mit Chrom(VI)-oxiddichlorid zu einer Formylgruppe oxidiert.[90.1]
Alkene und Alkine
[Bearbeiten | Quelltext bearbeiten]Durch Hydratisierung von Alkinen werden Enole gebildete, die sich zu Aldehyden oder Ketonen umlagern. Aldehyde speziell werden durch die Anti-Markovnikov-Hydratisierung terminaler Alkine erhalten, was mittels Hydroborierung und anschließender Oxidation möglich ist.[84.2] Die Wacker-Oxidation von Ethylen zu Acetaldehyd ist ein wichtiger industrieller Prozess. Mit anderen terminalen Alkenen werden meist bevorzugt Methylketone gebildet, mit Styrol als Ausnahme, das überwiegend zu Phenylacetaldehyd oxidiert wird.[91] Die Wacker-Oxidation nutzt Sauerstoff in Gegenwart von Palladium(II)-chlorid und Kupfer(II)-chlorid, normalerweise tritt Markovnikov-Selektivität zu Methylketonen auf; inzwischen sind jedoch Abwandlungen der Reaktionsbedingungen entwickelt worden, die in bestimmten Fällen eine umgekehrte Selektivität erzielen, was die Herstellung von Aldehyden ermöglicht.[92]
Oxidative Spaltung von Alkenen und Glycolen
[Bearbeiten | Quelltext bearbeiten]Bei Glycolspaltungen können je nach genutztem Oxidationsmittel Aldehyde oder Carbonsäuren entstehen. Aldehyde können beispielsweise unter Nutzung von Blei(IV)-acetat gebildet werden. Durch eine kombinierte Dihydroxylierung / Glycolspaltung können Alkene in Aldehyde / Carbonsäuren gespalten werden. Aldehyde können so in einer Reaktion mit Natriumperiodat und katalytisch Osmiumtetroxid gewonnen werden. Wird Osmiumtetroxid jedoch durch Kaliumpermanganat ersetzt oder durch Rutheniumtetroxid (Einsatz von katalytisch Ruthenium(III)-chlorid oder Ruthenium(IV)-oxid), werden Carbonsäuren erhalten, da diese Reagenzien die zunächst entstehenden Aldehyde weiter oxidieren.[93.1] Bei der Ozonolyse von passend substituierten Alkenen werden Aldehyde erhalten, wenn die Aufarbeitung mit einem milden Reduktionsmittel erfolgt, beispielsweise elementares Zink / Essigsäure, Triphenylphosphin oder Dimethylsulfid.[93.2]
Die Grundmann-Aldehyd-Synthese ist eine mehrstufige Umwandlung von Carbonsäurechloriden in Aldehyde. Das Carbonsäurechlorid wird zunächst mit Diazomethan homologisiert. Mit Essigsäure wird ein Acetat gebildet; Meerwein-Ponndorf-Verley-Reduktion und Hydrolyse ergibt ein Glycol. Glycolspaltung mit Blei(IV)-acetat ergibt dann ein Aldehyd mit der ursprünglichen Kettenlänge.[94.1]
Reduktion von Carbonsäurederivaten
[Bearbeiten | Quelltext bearbeiten]Bei der Reduktion von Carbonsäurederivaten kommt es leicht zur Überreaktion unter Bildung eines primären Alkohols. Eine verbreitete Methode, um dies zu vermeiden, ist die Reduktion eines Weinreb-Amids. Dieses wird aus einem Carbonsäurederivat mit N,O-Dimethylhydroxylamin-Hydrochlorid gebildet. Wird ein Hydrid an ein Weinreb-Amid addiert, entsteht ein stabiles tetraedrisches Intermediat. Da zunächst kein Aldehyd ausgebildet wird, ist auch keine weitere Hydrid-Addition und Überreaktion möglich. Bei der wässrigen Aufarbeitung erfolgt dann die Hydrolyse zum Aldehyd, dabei wird auch das eingesetzte Reduktionsmittel zerstört, sofern es nicht vollständig reagiert hat. Die Reduktion eines Weinreb-Amids ist mit Lithiumaluminiumhydrid oder DIBAL möglich.[95] Tertiäre Amide, sowohl Weinreb-Amide als auch N,N-Dialkylamide, können schnell und in guter Ausbeute mit dem Schwartz-Reagenz zu Aldehyden reduziert werden.[96]
In der Rosenmund-Reduktion findet eine katalytische Hydrierung von Carbonsäurechloriden zu Aldehyden statt, als Katalysator dient Palladium auf Bariumsulfat, vergiftet mit Chinolin und Schwefel.[97.1] In der Fukuyama-Reduktion wird ein Thiolester mit Triethylsilan zum Aldehyd reduziert, als Katalysator dient Palladium auf Kohlenstoff.[98.1] Carbonsäuren können mit DIBAL zu Aldehyden reduziert werden, indem sie zuvor in situ mit Trimethylsilylchlorid zu einem Trimethylsilylester umgesetzt werden.[99] Bei der McFadyen-Stevens-Reduktion wird ein Aldehyd aus einem gemischten Hydrazid (Carbonsäurehydrazid / Sulfonsäurehydrazid) hergestellt. Unter Einwirkung einer Base wird ein Sulfinat abgespalten, es entsteht eine Azoverbindung. Durch homolytische Bindungsspaltung wird Stickstoff freigesetzt und durch Radikalrekombination ein Aldehyd gebildet.[100.1]
{kind=link}
{kind=link}
Nitrile können auf verschiedenen Wegen zu Iminen reduziert werden, deren Hydrolyse leicht Aldehyde ergibt. Diverse Hydride sind hierzu geeignet, wobei DIBAL bei weitem am häufigsten genutzt wird. Alternativen sind andere Aluminiumhydride, Borane sind im Allgemeinen ungeeignet. Eine klassische Methode, deren Nutzung nachgelassen hat, die aber nach wie vor eingesetzt wird, ist die Stephen-Reduktion. Dabei wird das Nitril mit Zinn(II)-chlorid und wasserfreiem Chlorwasserstoff in Diethylether zum Imin reduziert und dann hydrolysiert. Unter geeigneten Bedingungen ist auch eine Reduktion von Nitrilen zu Iminen mit Raney-Nickel als Katalysator möglich, wodurch wiederum Aldehyde zugänglich sind.[87.4]
Carbonylierung und Formylierung
[Bearbeiten | Quelltext bearbeiten]Die Hydroformylierung von Alkenen ist ein großtechnischer Prozess mit einer Produktionsmenge in der Größenordnung von 10 Millionen Tonnen pro Jahr. Dabei werden Alkene in Gegenwart eines geeigneten Katalysators mit Synthesegas (Kohlenstoffmonoxid / Wasserstoff) zu Aldehyden umgesetzt. Gängige Katalysatoren sind Cobaltcarbonylhydrid, andere verwandte Cobaltkomplexe oder Rhodium mit Triphenylphosphin.[101]
{kind=link}
{kind=link}
Primäre Alkylhalogenide und Tosylate können mittels Kaliumtetracarbonylferrat(II) (Collmanns Reagenz) carbonyliert werden. Dabei wird zunächst der organische Rest des Edukts an den Komplex gebunden, Kohlenstoffmonoxid insertiert, das Komplexanion unter Einwirkung von Säure protoniert und ein Aldehyd freigesetzt.[102]
Eine Formylierung von aromatischen Verbindungen ist auf verschiedenen Wegen möglich. Bei der Gattermann-Synthese erfolgt die Reaktion mit Cyanwasserstoff und Chlorwasserstoff in Gegenwart einer Lewis-Säure, beispielsweise Aluminiumchlorid, ein Spezialfall der Friedel-Crafts-Acylierung. Die Cyanogruppe bildet mit Chlorwasserstoff ein Elektrophil von nicht genau bekannter Struktur und stellt das Kohlenstoffatom für die Formylgruppe zur Verfügung. Elektrophile aromatische Substitution ergibt ein Iminiumion, nach Hydrolyse entsteht ein Aldehyd.[97.2] Die Formylierung von Phenolen gelingt mit Urotropin in der Duff-Reaktion, dabei entsteht zunächst ein Imin, dessen Hydrolyse dann den Aldehyd ergibt.[103.1] In der Bouveault-Aldehyd-Synthese wird eine Grignard-Verbindung mit einem disubstituierten Formamid unter Bildung eines Aldehyds formyliert. Ein Beispiel ist die Umsetzung von 2-Bromtoluol über 2-Methylphenylmagnesiumbromid mit N-Methylformanilid zu 2-Methylbenzaldehyd. Eng verwandt ist die Bodroux-Tschitschibabin-Aldehydsynthese, bei der eine Grignard-Verbindung mit einem Orthoformiat umgesetzt wird.[103.2]
Umlagerungen
[Bearbeiten | Quelltext bearbeiten]Bei der Meyer-Schuster-Umlagerung werden Propargylalkohole durch Umsetzung mit einer Säure zu α,β-ungesättigten Aldehyden (im Falle eines terminalen Alkins) oder Ketonen umgelagert.[86.2] Allylalkohole können mittels Chloro(cyclopentadienyl)bis(triphenylphosphin)ruthenium oder Tetrapropylammoniumperruthenat zu Aldehyden umgelagert werden.[104] Epoxide können unter Einwirkung einer Lewis-Säure zu Aldehyden umgelagert werden (Meinwald-Umlagerung), beispielsweise 2-Phenylpropylenoxid mit Kupfer(II)-tetrafluoroborat zu 2-Phenylpropanal.[105.1]
Reaktionen
[Bearbeiten | Quelltext bearbeiten]Chemische Eigenschaften der Aldehyde
[Bearbeiten | Quelltext bearbeiten]{kind=link}
{kind=link}
Die C=O-Bindung der Carbonylgruppe in Aldehyden ist stark polar mit einer positiven Partialladung (δ+) am Kohlenstoffatom und negativer Partialladung (δ−) am Sauerstoffatom. Polare Reagenzien addieren gemäß Ladungsverteilung and die Carbonylgruppe: Nucleophile reagieren am Kohlenstoffatom, Elektrophile am Sauerstoffatom.[6.1] Andererseits beobachtet man, dass Wasserstoffatome am zur Carbonylgruppe benachbarten C-Atom deutlich acider sind als Wasserstoffatome an „normalen“ C-Atomen. Dies liegt zum einen daran, dass der Carbonylkohlenstoff sehr elektronenarm ist und einen −I-Effekt auf benachbarte Bindungen ausübt, zum anderen kann nach Deprotonierung die negative Ladung auf den Sauerstoff der Carbonylgruppe delokalisiert werden (−M-Effekt).[106] Aldehyde mit einem Wasserstoffatom, gebunden an das α-Kohlenstoffatom direkt neben der Carbonylgruppe, können in der Keto- und der Enolform vorliegen – siehe dazu Keto-Enol-Tautomerie.[107]
Oligomere und Polymere
[Bearbeiten | Quelltext bearbeiten]Verschiedene Aldehyde bilden Oligomere oder Polymere. Formaldehyd polymerisiert zu Paraformaldehyd beziehungsweise Polyoxymethylen. Unter Erhitzung wandelt sich Paraformaldehyd in 1,3,5-Trioxan um. Auch andere Aldehyde wie Acetaldehyd und Isobutyraldehyd können in Gegenwart von Säure trimerisieren.[108] Die Polymerisation von Formaldehyd zu hochmolekularem Polyoxymethylen gelingt anionisch (beispielsweise mit Bortrifluorid oder Perchlorsäure als Katalysator) oder anionisch (beispielsweise mit Tributylamin, Triphenylphosphin oder Diphenylzink als Katalysator).[109.1] Acetaldehyd kann zu Polyacetaldehyd polymerisiert werden, Trifluoracetaldehyd zu Polyfluoral, Trichloracetaldehyd zu Polychloral. Letztere beide sind chemisch extrem beständige Verbindungen.[109.2] Oligomere des Acetaldehyds sind Paraldehyd (Trimer), das sich unter sauren Bedingungen bildet. Metaldehyd (Tetramer) entsteht nur unter bestimmten Bedingungen, beispielsweise mit trockenem Chlorwasserstoff unter 0 °C.[110]
Hydrate, Acetale und Thioacetale
[Bearbeiten | Quelltext bearbeiten]Aldehyde stehen in wässriger Lösung mit dem entsprechenden Aldehydhydrat (gem-Diol), das heißt einem Kohlenwasserstoff mit zwei Hydroxygruppen an einem Kohlenstoffatom, im Gleichgewicht. In der Regel liegt das Gleichgewicht auf der Seite des Aldehyds. Im Falle des Trichloracetaldehyds liegt das Gleichgewicht jedoch auf der Seite des geminalen Diols.[111] Auch Formaldehyd liegt in wässriger Lösung zu 99,99 % als Aldehydhydrat vor, bei Acetaldehyd sind es 58 %.[112]
Cyclische Acetale werden beispielsweise mit Ethylenglycol, 1,3-Propandiol oder 2,2-Dimethyl-1,3-propandiol gebildet. Die Hydrolyse ist im Fall der 1,3-Dioxane (aus 1,3-Propandiol) deutlich schneller, die Bildung am langsamsten.[113.1] Die wichtigste Schutzgruppe insgesamt sind 1,3-Dioxolane, also cyclische Acetale von Ethylenglycol. Einführung der Schutzgruppe gelingt mit Ethylenglycol und einer Säure am Rückfluss, wobei entstehendes Wasser mit einem Wasserabscheider entzogen wird. Falls die Hitze ein Problem darstellt, kann Wasser auch durch ein wasserentziehendes Reagenz entfernt werden, beispielsweise Trimethylorthoformiat.[113.2] Acyclische Dithioacetale werden durch Reaktion mit einem Thiol (Methanthiol, Ethanthiol...) unter sauer Katalyse hergestellt.[113.3] Cyclische Dithioacetale werden mit 1,2-Ethandithiol oder 1,3-Propandithiol hergestellt, oft unter Lewis-Säure-Katalyse, beispielsweise mit Bortifluorid-Diethyletherat.[113.4]
Analog zur Bildung von Acetalen mit Alkoholen können Aldehyde Dithioacetale (S,S-Acetale) bilden, indem sie mit Thiolen beziehungsweise Dithiolen reagieren. Symmetrische Dithioacetale werden problemlos erhalten, indem Aldehyde und Thiole unter Säurekatalyse zur Reaktion gebracht werden. Unsymmetrische Vertreter, die Gruppen aus zwei unterschiedlichen Thiolen enthalten, sind schwieriger herzustellen. Sind die Thiole in ihren Eigenschaften ausreichend verschieden, ist jedoch eine vergleichsweise selektive Reaktion zu unsymmetrischen Acetalen möglich, indem Camphersulfonsäure als Katalysator eingesetzt wird.[114] Einfache, symmetrische Dithioacetale sind wichtige Intermediate in diversen Umsetzungen von Aldehyden. Dazu gehören beispielsweise die Takeda-Olefinierung (siehe Abschnitt Alkene), die Thioketal-Reduktion (siehe Abschnitt Alkane) und die Umpolung beziehungsweise Corey-Seebach-Reaktion (siehe unten).
Aldehyde können über ein Dithioacetal in Ketone überführt werden. Dabei wird der Aldehyd zunächst mit 1,3-Propandithiol umgesetzt. Das entstandene Thioacetal kann mittels Butyllithium deprotoniert werden und wird zu einem guten Nucleophil, im Gegensatz zum Aldehyd selbst, der ein Elektrophil ist. Man spricht von einer Umpolung, diese ist das Prinzip der Corey-Seebach-Reaktion. Umsetzung mit Alkylhalogeniden und Entfernung der Dithioacetalgruppe ergibt ein Keton. Zur Spaltung des Thioacetals eignen sich beispielsweise Quecksilber(II)-chlorid oder N-Bromsuccinimid.[115.1] In einer zweistufigen Reaktion können auch Cycloalkanone hergestellt werden, beispielsweise Cyclobutanon aus Formaldehyd (das umgepolt wird) und 1-Brom-3-chlorpropan. Durch Reaktion mit Kohlenstoffdioxid wird eine α-Oxocarbonsäure erhalten.[115.2]
Imine, Hydrazone und andere Stickstoffderivate
[Bearbeiten | Quelltext bearbeiten]Aldehyde kondensieren mit primären Aminen (beispielsweise N-Butylamin) zu Iminen, wenn dem Reaktionsgemisch Wasser entzogen wird. Mit sekundären Aminen (beispielsweise Piperidin) bilden sich stattdessen Enamine. In Ausnahmefällen, wenn ein Aldehyd keine α-Wasserstoffatome hat, können sich Aminale bilden, beispielsweise bei der Reaktion von Benzaldehyd mit Piperidin. Mit anderen Stickstoffverbindungen können zu Iminen analoge Verbindungen synthetisiert werden, beispielsweise Oxime aus Hydroxylamin oder Hydrazone aus Hydrazinen.[116][116.1]
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Neben der Kondensation kann auch die Aza-Wittig-Reaktion zur Herstellung von Iminen dienen. Dabei wird ein Iminophosphoran wie N-Phenyliminotriphenylphosphoran mit einer Carbonylverbindung umgesetzt, als Nebenprodukt fällt ein Phosphanoxid an.[86.3]
{kind=link}
{kind=link}
Imine und Iminiumsalze können mit diversen Reagenzien zu Aminen reduziert werden. Natriumcyanoborhydrid ist insofern besonders, dass es bei geeignetem pH-Wert praktisch nur Iminiumverbindungen reduziert. Dadurch ermöglicht es die Eintopfsynthese von primären, sekundären und tertiären Aminen aus einem Aldehyd mit Ammoniak, einem primären oder sekundären Amin (reduktive Aminierung). Dabei wird in situ aus Aldehyd und Ammoniak / Amin eine Iminiumverbindung gebildet und diese reduziert, während Natriumcyanoborhydrid mit dem Aldehyd nicht interagiert. Die reduktive Aminierung ist eine zuverlässige Methode mit breitem Anwendungsfeld.[117.1] Eine Namensreaktion, die ebenfalls der Umwandlung von Aldehyden in Amine dient, ist die Leuckart-Wallach-Reaktion, bei der der Aldehyd mit einem Amin plus Ameisensäure (als Reduktionsmittel) oder schlicht mit Ammoniumformiat umgesetzt wird.[105.2] Ein Spezialfall ist Eschweiler-Clarke-Reaktion, bei der ein primäres oder sekundäres Amin mit Formaldehyd und Ameisensäure methyliert wird. Im Falle eines primären Amins ist so eine selektive Gewinnung von tertiären N,N-Dimethylaminen möglich.[88.2]
{kind=link}
{kind=link}
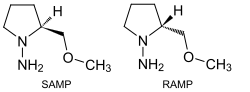
Im Gegensatz zu Ketonen und Estern werden Aldehyde selten in Enolate überführt, um diese zu alkylieren, da die Zugabe von Base typischerweise zu Aldolreaktionen führt. Dies kann vermieden werden, wenn die Enolatbildung zügig und quantitativ erfolgt, was beispielsweise mit Kaliumhydrid in Tetrahydrofuran oder mit Kaliumamid in flüssigem Ammoniak möglich ist. Allgemein können solche Reaktionen jedoch durchgeführt werden, indem Aldehyde zunächst in ein Enamin oder ein Enamin-Anion (durch Deprotonierung eines Imins) überführt werden.[118] Das Enders-Reagenz, (S)- oder (R)-1-Amino-2-methoxymethylpyrrolidin (auch SAMP und RAMP), ist ein wichtiges chirales Auxiliar. Mit der Verbindung können Aldehyde in die entsprechenden Hydrazone überführt werden. Metallierung ergibt ein nucleophiles Azaenolat, das in α-Stellung mit Elektrophilen modifiziert werden kann. Die Struktur des Auxiliars ermöglicht dabei eine hohe Stereoselektivität. Eine große Rolle spielt es insbesondere bei stereoselektiven Alkylierungen von Aldehyden und Ketonen, aber auch für Aldoladditionen oder Michael-Additionen.[119]
Die eher selten genutzte Schützung in Form eines Hydrazons nutzt beispielsweise N,N-Dimethylhydrazin, Phenylhydrazin oder para-Toluolsulfonsäurehydrazid.[113.5]
Aldolreaktionen
[Bearbeiten | Quelltext bearbeiten]Bei der Aldoladdition wird das CH-acide Wasserstoffatom in der α-Position durch eine Base abgespalten. Das entstandene Enolatanion addiert an den Carbonylkohlenstoff eines weiteren Aldehyd-Moleküls. Es entsteht ein Aldol, ein Additionsprodukt aus Alkohol (OH-Gruppe) und Aldehyd. Auf diese Weise können C-C-Bindungen geknüpft werden. Wird das gebildete Aldol anschließend dehydratisiert, spricht man von Aldolkondensation, dabei entstehen α,β-ungesättigte Aldehyde.[111] Zwei Moleküle des gleichen Aldehyds können leicht verknüpft werden. Bei gekreuzten Aldolreaktionen werden zwei unterschiedliche Carbonylverbindungen eingesetzt werden, die jeweils als Nucleophil oder Elektrophil wirken können. Dabei kann es schwierig sein, gezielt ein Produkt zu gewinnen, da insgesamt vier Produkte entstehen können. Ist ein Reaktionspartner ein Aldehyd ohne α-Wasserstoffatome, beispielsweise Benzaldehyd fällt dieser Effekt weniger stark aus, da nur zwei Produkte möglich sind, aus dem enolisierbaren Aldehyd mit sich selbst oder mit dem anderen Aldehyd, und das gekreuzte Produkt ist häufig das Hauptprodukt. Eine solche Reaktion wird als Claisen-Schmidt-Kondensation bezeichnet. Unter geeigneten Bedingungen können aber mit allen Aldehyden gezielte Aldolreaktionen durchgeführt werden. Indem ein Aldehyd mit einer Amidbase, beispielsweise bei −78 °C mit Lithiumdiisopropylamid in Tetrahydrofuran, umgesetzt wird, wird er weitgehend deprotoniert. Wird erst dann der zweite Aldehyd zugegeben, ist eine selektive Reaktion möglich. Auch Reaktionen zwischen Aldehyd und Keton sind möglich, einerseits in einer Claisen-Schmidt-Reaktion mit Keton und nicht enolisierbarem Aldehyd oder in der Variante mit einer Amidbase, bei der eines oder beide der Edukte gegen ein Keton ausgetauscht werden können.[120] In der Nitroaldol-Reaktion wird ein Nitroalkan deprotoniert und an ein Aldehyd oder Keton addiert. Durch Wasserabspaltung entstehen Nitroalkene.[121]
Alkohole: Reaktionen mit Wasserstoff und Hydriden
[Bearbeiten | Quelltext bearbeiten]Mit geeigneten Reduktionsmitteln wie Natriumborhydrid können Aldehyde zu primären Alkoholen reduziert werden. Als Lösungsmittel für diese Reaktion eignen sich Wasser und Alkohole wie Methanol und Ethanol. Aldehyde lassen sich leichter reduzieren als Ketone, sodass Natriumborhydrid in Gegenwart eines Ketons selektiv einen Aldehyd reduzieren kann.[122] Die umgekehrte Selektivität wird in der Luche-Reduktion erzielt, bei der ein Keton mit Cer(III)-chlorid aktiviert wird und dann in Gegenwart eines Aldehyds selektiv mit Natriumborhydrid reduziert werden kann.[123] Natriumborhydrid wird bei weitem am meisten genutzt, diverse andere komplexe Borhydride finden aber ebenfalls Anwendung zur Reduktion von Aldehyden: Kaliumborhydrid hat ähnliche Eigenschaften wie Natriumborhydrid. Zinkborhydrid, das in situ aus Natriumborhydrid und Zinkchlorid hergestellt werden kann, kann selektiv α,β-ungesättigte Aldehyde reduzieren und die chelatisierenden Eigenschaften des Zinks verbessern die Diastereoselektivität, wenn ein Aldehyd eine Hydroxy- oder Alkoxygruppe trägt. Quartäre Ammoniumsalze wie Tetrabutylammoniumborhydrid sind etwas weniger reaktiv als Natriumborhydrid, sind aber sehr stabil und einfach in der Handhabung.[117][117.2] Ein hochselektives Reagenz, das Aldehyde sehr leicht, Ketone jedoch nur in Ausnahmen reduziert, ist das Natriumtriacetoxyborhydrid.[124] Das hochreaktive Lithiumaluminiumhydrid reagiert schon bei Raumtemperatur schnell und im Wesentlichen quantitativ mit den meisten Carboxyl- und Carbonylverbindungen inklusive Aldehyden.[125]
Aldehyde können außerdem zu primären Alkoholen hydriert werden, beispielsweise unter Katalyse mit Mangan, Rhenium oder Ruthenium.[126][127]
In der Meerwein-Ponndorf-Verley-Reduktion wird ein Aldehyd durch Reaktion mit Aluminiumisopropanolat reduziert. Dabei werden die Isopropanolat-Gruppen zu Aceton oxidiert. Es handelt sich um eine Gleichgewichtsreaktion, jedoch kann das Gleichgewicht verschoben werden, indem während der Reaktion das entstehende Aceton abdestilliert wird.[128]
Alkohole: Reaktionen mit Organometallverbindungen und andere
[Bearbeiten | Quelltext bearbeiten]Die Addition von Grignard-Reagenzien an Aldehyde ergibt ebenfalls Alkohole. Im Fall von Formaldehyd entsteht dabei ein primärer Alkohol, bei anderen Aldehyden ein sekundärer Alkohol.[129] In der Reformatzki-Reaktion werden α-halogenierte Carbonsäureverbindungen mit elementarem Zink umgesetzt und an Aldehyde addiert, wodurch eine Alkoholgruppe in einem β-Hydroxyderivat gebildet wird.[130] Durch Addition von Allyltributylzinn, meist unter Katalyse einer Lewis-Säure, können aus Aldehyden Homoallylalkohole hergestellt werden.[131] In der Nozaki-Hiyama-Kishi-Reaktion wird ein eine Organohalogenverbindung (Aryl-, Allyl-...) unter Katalyse mit Nickel(II)-chlorid mit Chrom(II)-chlorid zu einer Organochromverbindung umgesetzt, die dann an einen Aldehyd addiert werden kann, um Allylalkohole, Homoallylalkohole und so weiter herzustellen.[132] Bei der Soai-Reaktion wird Diisopropylzink an einen Pyrimidin-Aldehyd addiert, beispielsweise an Pyrimidin-5-carbaldehyd, wobei als Besonderheit durch Autokatalyse und Enantioselektivität eine Amplifikation eines kleinen Ausgangs-Enantiomerenüberschusses zu deutlich höheren Werten auftritt.[133]
In der Pinakol-Kupplung bilden zwei Aldehyde eine C-C-Bindung aus, wodurch ein Diol entsteht. Setzt man Aldehyde mit einem Alkalimetall (Beispiel: Natrium) um, so bildet sich ein Radikal-Anion, das schnell dimerisiert. Die Hydrolyse liefert ein Pinakol (traditionelle Bezeichnung für ein 1,2-Diol, also ein Diol mit vicinalen Hydroxygruppen). Ausgehend von einem α,ω-Dialdehyd erhält man analog durch eine intramolekulare Reaktion cyclische 1,2-Diole.[134]
Carbonsäuren und Carbonsäurederivate
[Bearbeiten | Quelltext bearbeiten]Aldehyde sind reaktive Verbindungen und lassen sich sehr leicht zur Carbonsäure oxidieren.[111] Eine Autoxidation von Aldehyden zu Carbonsäuren durch Luftsauerstoff tritt häufig auf, hat aber keine synthetische Bedeutung. Historische Oxidationsmethoden sind die nach Fehling mit Kupfer(II) oder nach Tollens mit Silber(I). Die Jones-Oxidation mit Chrom(VI) wird nach wie vor genutzt. Neuere Methoden sind die nach Lindgren und ihre Weiterentwicklung nach Pinnick, die beide Natriumchlorit als Oxidationsmittel nutzen.[135][135.1] Bei der Delépine-Reaktion werden Aldehyde mit Silber(I)-oxid in Natronlauge zu Carbonsäuren oxidiert.[94.2] Geeignete Reagenzien sind zum Beispiel Silber(I)-oxid (das entstehende elementare Silber dient dem Nachweis von Aldehyden in der Silberspiegelprobe) oder Peroxycarbonsäuren wie meta-Chlorperbenzoesäure.[136]
In der Tischtschenko-Reaktion werden Aldehyde zu symmetrischen Carbonsäureestern dimerisiert. Beispielsweise kann Benzylbenzoat aus Benzaldehyd hergestellt werden. Als Katalysator kommen Aluminiumalkoholate wie Aluminiumtriethanolat zum Einsatz.[137] Die Corey-Gilman-Ganem-Oxidation ist eine Methode um α,β-ungesättigte Aldehyde zu oxidieren und direkt in entsprechende Carbonsäuremethylester umzusetzen. Dazu wird das Edukt in Methanol in Gegenwart von Cyanwasserstoff mit Mangan(IV)-oxid zur Reaktion gebracht. Es entsteht ein Cyanhydrin, das zu einem Acylcyanid oxidiert wird und dann mit Methanol zum Ester reagiert.[138]
Durch die Reaktion mit tert-Butylhypochlorit können Aldehyde direkt in Carbonsäurechloride überführt werden.[139] In Gegenwart von Natriumazid können so auch Carbonsäureazide hergestellt werden.[140] Durch eine radikalische Reaktion mit Ethyltribromacetat und Dibenzoylperoxid können Aldehyde in Carbonsäurebromide umgewandelt werden,[141] durch Reaktion mit Trichlorisocyanursäure und Caesiumfluorid können Aldehyde in Carbonsäurefluoride umgewandelt werden.[142]
Die Herstellung von Nitrilen aus Aldehyden verläuft in der Regel über Aldoxime (siehe Abschnitt Herstellung von Iminen und verwandten Verbindungen). Diese werden durch Reaktion des Aldehyds mit Hydroxylaminhydrochlorid hergestellt und anschließend beispielsweise mit Oxalylchlorid dehydratisiert.[143] Die direkte Umsetzung von Aldehyden zu Nitrilen ist unter anderem mit Hydroxylamin-O-sulfonsäure[144] möglich oder indem die Reaktion mit Hydroxylamin in Gegenwart von Titan(IV)-chlorid durchgeführt wird.[145]
Die Cannizzaro-Reaktion ist die Disproportionierung eines nicht-enolisierbaren Aldehyds zu einem Alkohol und einem Carboxylat. Im ursprünglichen Beispiel von Cannizzaro ergab die Reaktion von Benzaldehyd mit Kaliumhydroxid als Produkte Benzylalkohol und Kaliumbenzoat.[146]
Alkane
[Bearbeiten | Quelltext bearbeiten]Durch einige Methoden kann die Aldehyd-Funktion zu einer Methylgruppe reduziert werden. Eine solche Methode ist die Wolff-Kishner-Reduktion. Hierzu muss der Aldehyd in ein Hydrazon umgewandelt werden, dies ist jedoch auch in situ möglich. In dem Fall wird das Edukt mit Hydrazinhydrat und einer Base (Natriumhydroxid oder Kaliumhydroxid) in einem hochsiedenden Lösungsmittel (beispielsweise Diethylenglycol) für mehrere Stunden auf 180 °C bis 200 °C erhitzt.[147] Eine verwandte Methode ist die Clemmensen-Reduktion, bei der die Aldehydgruppe mit Zinkamalgam und Salzsäure reduziert wird. Wolff-Kishner-Reduktion und Clemmensen-Reduktion sind komplementär, da im einen Fall im Basischen und im anderen im Sauren gearbeitet wird.[148] Eine weitere Methode, die Thioketal-Reduktion, verläuft über die Thioacetale, meist mit Ethandithiol. Diese können durch diverse Reduktionsmittel reduziert werden, ursprünglich mit Raney-Nickel. Andere geeignete Reagenzien sind Komplexe aus Nickel(II)-acetat, Natriumhydrid und tert-Amylalkohol sowie Dinickelborid, das aus Nickel(II)-chlorid und Natriumborhydrid hergestellt wird.[149] Weitere Methoden verwenden Amminboran mit Titanocendichlorid und Lithiummethanolat[150] oder Polymethylhydrosiloxan mit Tris(pentafluorphenyl)boran.[151]
Durch Decarbonylierung von Aldehyden wird ebenfalls eine Kohlenwasserstoff-Verbindung erhalten. Durch die Abspaltung von Kohlenmonoxid besitzt diese jedoch ein Kohlenstoffatom weniger als das Edukt. Die Decarbonylierung ist beispielsweise durch Reaktion mit dem Wilkinson-Katalysator, einem Rhodium-Komplex, möglich.[152] Mit Dichlordimethyltitan können aromatische Aldehyde zweifach methyliert werden.[153]
Alkene
[Bearbeiten | Quelltext bearbeiten]Die Wittig-Reaktion ist eine sehr bedeutende Methode der organischen Synthese, bei der ein Aldehyd oder Keton mit einem Phosphor-Ylid zu einem Alken umgesetzt wird. Ein Aldehyd kann mit dem Phosphor-Ylid ein Oxaphosphetan, einen Vierring mit P-O-Bindung, als Zwischenstufe bilden. Dieses zerfällt dann in ein Alken und Triphenylphosphanoxid. Einen wichtigen Beitrag zur Triebkraft der Reaktion stellt die Ausbildung einer Phosphor-Sauerstoff-Bindung.[154] Die nötigen Ylide werden allgemein ausgehend von Triphenylphosphin und einem Alkylhalogenid hergestellt. Durch nukleophile Substitution wird zunächst ein Phosphoniumsalz hergestellt. Die Deprotonierung mit einer starken Base, oft eine Organolithium-Verbindung, ergibt dann das Ylid.[155] Eine abgeleitete Methode, mit der ebenfalls Aldehyde zu Alkenen umgesetzt werden, ist die Horner-Wadsworth-Emmons-Reaktion. Hier ist das Reagenz ein Phosphanoxid mit zwei Phenylgruppen und einer Alkylgruppe, die übertragen wird. Dieses wird beispielsweise mit Butyllithium zu einem Carbanion deprotoniert. Das Nebenprodukt ist ein Salz der Diphenylphosphinsäure.[156] Eine wesentlich wichtigere Weiterentwicklung ist die Horner-Wadsworth-Emmons-Reaktion (HWE-Reaktion). Hier ist das Reagenz ein Phosphonat, das mittels Arbusow-Reaktion hergestellt werden kann und durch Deprotonierung ebenfalls ein Carbanion bildet. Als Base eignen sich Natriumhydrid, Kaliumhydrid, Lithium-, Natrium- und Kaliumhexamethyldisilazid oder Kalium-tert-butanolat. Gegenüber der ursprünglichen Wittig-Reaktion hat diese Methode insbesondere den Vorteil, dass als Nebenprodukt ein Phosphorsäureester entsteht, der im Gegensatz zum Triphenylphosphanoxid wasserlöslich ist und leicht entfernt werden kann.[157]
In der Julia-Lythgoe-Olefinierung kommt als Olefinierungsreagenz ein Sulfon zum Einsatz. Dieses wird mit einer Organometallverbindung wie Butyllithium deprotoniert und kann dann einen Aldehyd unter Bildung einer C-C-Einfachbindung angreifen. Die aus dem Aldehyd gebildete Alkoholat-Gruppe wird acyliert, beispielsweise mit Acetanhydrid. Im letzten Schritt wird mit einem Reduktionsmittel wie Natriumamalgam eine reduktive Eliminierung durchgeführt, wobei Sulfon- und Estergruppe entfernt werden und trans-selektiv eine C=C-Doppelbindung gebildet wird.[158] Bei der verwandten Julia-Kociensky-Olefinierung wird ein Sulfon verwendet, das eine Arylgruppe mit Elektronenakzeptor-Eigenschaften trägt, beispielsweise Benzothiazol. Wird ein solches Sulfon deprotoniert und mit einem Aldehyd umgesetzt, sind keine weiteren Reaktionsschritte nötig. Es kommt zu einer Umlagerung der Arylgruppe auf das Sauerstoffatom, das zu Aldehyd gehörte und diese Gruppe wird durch Eliminierung abgespalten. Daneben entsteht Schwefeldioxid.[159] In der Peterson-Olefinierung wird das α-Carbanion einer Silylverbindung an einen Aldehyd addiert. Durch Eliminierung bildet sich ein Alken und ein Silanol, das Sauerstoffatom wird auf das Siliciumatom übertragen.[160]
Für die Methylenierung von Aldehyden, das heißt den Austausch des Sauerstoff-Atoms gegen eine CH2-Gruppe sind eine große Zahl an Reagenzien beziehungsweise Reaktionen bekannt. Besonders bekannt ist das Tebbe-Reagenz.[161][162] Verwandt mit dem Tebbe-Reagenz beziehungsweise davon abgeleitet sind das Grubbs-Reagenz und das Petasis-Reagenz.[162] Bei der Takai-Lombardo-Reaktion wird Dibrommethan mit elementarem Zink und Titan(IV)-chlorid verwendet. Ein Carben des Molybdäns, das aus Molybdän(V)-chlorid und Methyllithium hergestellt wird, ist besonders selektiv für Aldehyde.[161]
Eine neuere Method ist die Takeda-Olefinierung. Dabei wird ein Dithioacetal mit einem Derivat des Titanocens zu einem Carbenkomplex umgesetzt. Dieser reagiert dann mit einer Carbonylgruppe, beispielsweise in einem Aldehyd, unter Ausbildung einer C=C-Doppelbindung. Auch das Dithioacetal kann aus einem Aldehyd hergestellt werden.[162] Mit der McMurry-Reaktion können zwei Aldehyd-Moleküle durch Reaktion mit Titan(III)-chlorid direkt über eine C=C-Doppelbindung verbunden werden.[163]
Alkine
[Bearbeiten | Quelltext bearbeiten]Es sind mehrere Reaktionen bekannt, durch die aus Aldehyden Alkine hergestellt werden können, wobei jeweils ein zusätzliches Kohlenstoffatom eingeführt wird. In der Corey-Fuchs-Reaktion wird aus dem Aldehyd ein gem-Dibromolefin hergestellt, indem es mit Triphenylphosphin und Tetrabrommethan umgesetzt wird. Reaktion mit Butyllithium ergibt ein Lithiumacetylid, aus dem durch Hydrolyse ein terminales Alkin entsteht. Alternativ kann das Lithiumaceylid durch Reaktion mit einem Elektrophil wie Kohlenstoffdioxid, einem Alkylhalogenid, einem weiteren Aldehyd oder einem Epoxid weiter modifiziert werden.[164][165][166]
Bei der Seyferth-Gilbert-Homologisierung wird das zusätzliche Kohlenstoffatom mittels Dimethyldiazomethylphosphonat (DAMP) eingeführt. Als Base kommt hier Kalium-tert-butanolat zum Einsatz. Bei der Weiterentwicklung nach Ohira und Bestmann wird das Ohira-Bestmann-Reagenz eingesetzt. Aus diesem wird durch Reaktion mit Kaliumcarbonat in situ DAMP hergestellt.[164]
Heterocyclen
[Bearbeiten | Quelltext bearbeiten]Aldehyde sind häufige Edukte für die Synthese von Heterocyclen. Dazu gehört beispielsweise die Herstellung von Pyridinen aus Ammoniak und Aldehyden mittels Chichibabin-Reaktion[167] oder aus Ammoniak, einem Aldehyd und einer Dicarbonylverbindung in der Hantzsch-Pyridinsynthese.[168] Aldehyde dienen als Edukte in der Herstellung von Chinolinen mittels Friedländer-Chinolin-Synthese.[169] In der Skraup-Synthese wird Chinolin aus Anilin und Glycerin gebildet. Letzteres wird in situ in Acrolein umgewandelt. Die Doebner-Miller-Reaktion ist eine Verallgemeinerung, in der andere Glycole oder ungesättigte Aldehyde eingesetzt werden, um substituierte Chinoline zu erzeugen.[170] In der Paal-Knorr-Synthese können 1,4-Dicarbonylverbindungen (sowohl Ketone als auch Aldehyde) zu Furanen cyclisiert werden.[171] Mit Abwandlungen kann die Methode auch zur Herstellung von Thiophenen und Pyrrolen dienen. Erstere können gebildet werden, wenn Phosphorpentasulfid oder Lawessons Reagenz eingesetzt wird,[172] letztere, wenn ein primäres Amin eingesetzt wird.[173] Imidazole können aus einem Imidoester und einem Aminoaldehyd (aus Aminosäuren mittels Akabori-Reduktion) gewonnen werden, wobei beide Verbindungen zunächst zu einem Amidin reagieren und dann cyclisieren.[174] In der Asinger-Reaktion werden aus zwei Molekülen einer Carbonylverbindung (Keton oder Aldehyd) sowie Ammoniak und Schwefel Thiazoline gebildet.[175] In der Corey-Chaykovsky-Reaktion wird Dimethylsulfoniummethylid oder Dimethylsulfoxoniummethylid unter Bildung eines Dreirings an eine Doppelbindung addiert, im Falle der C=O-Doppelbindung eines Aldehyds wird so ein Epoxid erhalten.[176]
Chalkogen-Analoga, Bisulfit-Addukte und Halogenide
[Bearbeiten | Quelltext bearbeiten]Mit geeigneten Reagenzien können Aldehyde direkt in ihre Analoga mit den höheren Chalkogenen umgewandelt werden. Die Umwandlung in Selenoaldehyde gelingt mit Bis(trimethylsilyl)selenid in Gegenwart einer katalytischen Menge Butyllithium. Die Triebkraft ist die Ausbildung einer Silicium-Sauerstoff-Bindung. Die gebildeten Selenoaldehyde sind sehr instabil, können aber in einer Diels-Alder-Reaktion mit Cyclopentadien abgefangen werden. Eine analoge Herstellung von Thioaldehyden gelingt mit Bis(trimethylsilyl)sulfid.[177] Telluroaldehyde können mittels Bis(dimethylaluminium)tellurid hergestellt werden.[178]
{kind=link}
{kind=link}
In der Bisulfit-Reaktion mit Natriumhydrogensulfit bilden Aldehyde wasserlösliche Addukte, die von einem Gemisch abfiltriert werden können. Durch Reaktion mit Natriumcarbonat oder Salzsäure können die Aldehyde dann zurückgewonnen werden.[179] Die Addukte sind strukturell die α-hydroxylierten Derivate von Sulfonsäuren.[180]
Mit Diethylaminoschwefeltrifluorid können Aldehyde in geminale Difluoride umgewandelt werden,[181] mit 2,2,2-Tribrom-1,3,2-benzodioxaphosphol in geminale Dibromide.[182]
Cyanhydrine
[Bearbeiten | Quelltext bearbeiten]Durch Addition von Cyanid (beispielsweise aus Natriumcyanid) im Sauren (beispielsweise in Essigsäure) können aus Aldehyden Cyanhydrine hergestellt werden.[183] Anderes Reagenzien zur Herstellung von Cyanhydrinen sind Diethylaluminiumcyanid[184] und Acetoncyanhydrin, dessen Cyanwasserstoff-Einheit auf andere Aldehyde und Ketone übertragen werden kann.[185] Cyanhydrine kommen als Intermediate bei der Stetter-Reaktion (siehe Abschnitt Herstellung von Carbonylverbindungen) und bei der Corey-Gilman-Ganem-Oxidation von Aldehyden (siehe Abschnitt Herstellung von Carbonsäuren und deren Derivaten) vor.
Eine weitere Alternative für die Schützung sind Derivate von Cyanhydrinen, beispielsweise Acetate herstellbar mit Acetoncyanhydrin / Acetanhydrid oder mit Acetylcyanid, sowie Silylether, beispielsweise herstellbar mit Trimethylsilylcyanid.[113.6]
Auch in der Strecker-Synthese wird Cyanwasserstoff beziehungsweise Cyanid an Aldehyde addiert, in diesem Fall in Gegenwart von Ammoniak. Dabei entsteht zunächst ein α-Aminonitril, das zu einer Aminosäure hydrolysiert werden kann. Aminosäuren sind sowohl in biologischen Prozessen als auch in der Industrie sehr wichtige Verbindungen, was die historische Bedeutung der Methode begründet. Allerdings wird die Reaktion nach wie vor verbreitet verwendet.[186]
In der Stetter-Reaktion werden Aldehyde an α,β-ungesättigte Ketone, Ester oder Nitrile addiert, wodurch eine entsprechende 4-Oxoverbindung erhalten wird. Katalysiert wird die Reaktion durch N-Heterocyclische Carbene, die durch Deprotonierung von Thiazolen und Imidazolen erhalten werden.[187] Alternativ kann auch Cyanid (beispielsweise als Natriumcyanid) als Katalysator eingesetzt werden, wodurch als Intermediat das Cyanhydrin des Aldehyds entsteht.[188]
Schutzgruppen für Aldehyde
[Bearbeiten | Quelltext bearbeiten]Die wichtigsten Schutzgruppen für Aldehyde sind Acetale, inklusive O,O-Acetale, Dithioacetale und O,S-Acetale, die cyclisch oder acyclisch sein können. Eingeführt werden sie durch Reaktion der zu schützenden Verbindung mit Alkoholen, Thiolen, Diolen und so weiter unter sauren Bedingungen. O,O-Acetale sind stabil gegen Basen, Organometallverbindungen und anderen Nucleophile. gegen Reduktion durch Hydride und gegen viele Oxidationsmittel. Dithioacetale sind hingegen gegen viele Oxidationsmittel empfindlich und werden durch sehr starke Basen wie Butyllithium deprotoniert. O,O-Acetale können leicht durch saure Hydrolyse entschützt werden, Dithioacetale können unter oxidativen Bedingungen entschützt werden oder mit geeigneten Metallsalzen (Quecksilber, Silber, Kupfer). Andere weniger genutzte Schutzgruppen sind beispielsweise Cyanhydrine und Hydrazone.[113][113.7]
Weiterhin können Aldehyde in Form verschiedener Addukte geschützt werden. Ein Diethylamin-Addukt kann in situ mit Titantetrakis(diethylamid) hergestellt werden, separate Reaktionsschritte für Schützung / Entschützung entfallen. Mit Pyrrollithium wird ein Pyrrol/Alkohol gebildet, die Hydroxygruppe kann zusätzlich als Silylether geschützt werden. Die Bildung von Bisulfit-Addukten mit Natriumhydrogensulfit eignet sich sowohl zur Schützung als auch zur Aufreinigung, da sich praktische kristalline Verbindungen bilden.[113.8]
Verwendung
[Bearbeiten | Quelltext bearbeiten]Aldehyde sind eine wichtige Verbindungsklasse in diversen Anwendungsbereichen. Viele andere Chemikalien werden ausgehend von Aldehyden hergestellt. Außerdem werden viele Aldehyde als Duftstoffe und Aromen verwendet. Eine mengenmäßig sehr wichtige Anwendung ist die Herstellung von Polymeren aus Formaldehyd.
Intermediate in der chemischen Industrie
[Bearbeiten | Quelltext bearbeiten]Die wichtigste Anwendung von Acetaldehyd ist als Zwischenprodukt bei der Herstellung anderer Chemikalien, unter anderem Pyridin und seine Derivate, sowie Pentaerythrit und Peressigsäure.[189] Pentaerythrit wird aus Acetaldehyd und Formaldehyd unter basischer Katalyse hergestellt.[190] Als Basen eignen sich beispielsweise Natriumhydroxid oder Calciumhydroxid.[1] Verschiedene Pyridine werden aus Acetaldehyd und Ammoniak sowie jeweils einem weiteren Aldehyd nach der Tschitschibabin-Pyridinsynthese hergestellt.[191] Eine Herstellungsmethode von Ethylacetat ist durch Tischtschenko-Reaktion von Acetaldehyd. Die Bedeutung von Acetaldehyd als chemisches Intermediat hat aber insgesamt nachgelassen; für einige Verbindungen, beispielsweise Essigsäure und Acetanhydrid, die früher aus Acetaldehyd hergestellt wurden, gibt es heute andere bevorzugte Verfahren, beispielsweise den Monsanto-Prozess für Essigsäure.[1] Formaldehyd ist ein sehr wichtiger C1-Baustein und dient unter anderem zur Herstellung von 1,4-Butandiol, Nitrilotriessigsäure und EDTA, Pentaerythrit und Methylendiphenyldiisocyanat.[192]
Butanal wird insbesondere zu 1-Butanol reduziert,[193] der wiederum zu Butylacrylat, Butylacetat und Weichmachern weiterverarbeitet wird oder als Lösungsmittel eingesetzt wird, z. B. in Farben.[194] Ein weiteres wichtiges Folgeprodukt von Butanal ist 2-Ethylhexanol.[193] 2-Ethylhexanol wird vor allem zu Weichmachern weiterverarbeitet.[195] Zu den 2-Ethylhxylestern, die als Weichmacher verwendet werden, gehören z. B. Bis(2-ethylhexyl)phthalat, Bis(2-ethylhexyl)terephthalat und Bis(2-ethylhexyl)adipat.[196] Neben Butanal werden auch diverse andere Aldehyde zur Herstellung von Alkoholen durch Reduktion verwendet. Aus Pentanalen hergestellte Pentanole dienen ebenfalls als Lösungsmittel sowie zur Herstellung von Weichmachern und anderen Estern. Auch längerkettige Aldehyde werden zu Weichmacheralkoholen umgesetzt, beispielsweise Isooctanal zu Isooctanol. Citral ist der Ausgangsstoff für die synthetische Herstellung von Vitamin A.[1] Phenylacetaldehyd wird zur Herstellung von Phenylalanin über die Strecker-Synthese verwendet, das wiederum zu dem Süßstoff Aspartam umgesetzt wird.[1] Eine weitere Aminosäure, die in großen Mengen hergestellt und insbesondere als Zusatz in Tierfutter verwendet wird, ist Methionin. Es wird durch Strecker-Synthese aus Acrolein, Schwefelwasserstoff und Cyanwasserstoff hergestellt.[197][198]
Kunststoffe
[Bearbeiten | Quelltext bearbeiten]Die Produktionsmenge von Formaldehyd betrug 2022 etwa 23 Millionen Tonnen. Der Hauptanwendungsbereich von Formaldehyd ist die Herstellung von Polymeren: Harnstoffharze (Aminoplaste), Phenolharze (Phenoplaste) und Polyoxymethylen (POM).[199] Aminoplaste werden überwiegend durch Polykondensation von Formaldehyd und Harnstoff hergestellt. Ihre Hauptanwendung ist als Klebstoff für die Herstellung von Spanplatten und Sperrholz.[200] Aus POM werden beispielsweise Bauteile für Autos hergestellt, um deren Gewicht zu reduzieren.[192]
Butanal wird in der Herstellung von Polyvinylbutyral eingesetzt.[193] Polyvinylbutyral wird durch Polymerisation von Vinylacetat zu Polyvinylacetat, Hydrolyse zu Polyvinylalkohol und anschließende Acetalbildung mit Butanal hergestellt. Verwendet wird es hauptsächlich als Zwischenschicht in Sicherheitsglas, die für Bruchfestigkeit und Splitterschutz sorgt. Verwendet wird solches Glas für diverse Anwendungen inklusive Autofenstern, Glastüren und Duschwänden.[201]
Aromen und Duftstoffe
[Bearbeiten | Quelltext bearbeiten]Acetaldehyd wird als Aromastoff verwendet.[202] Citronellal wird als Duftstoff beispielsweise in Seife und Waschmitteln eingesetzt. Citral wird selbst als Duftstoff eingesetzt, aber auch zu Acetalen weiterverarbeitet, die ebenfalls als Duftstoffe dienen. Längerkettige Alkanale wie Octanal werden insbesondere als Duftstoffe für Zitrusnoten verwendet. Phenylacetaldehyd hat einen Duft nach Rosen oder Hyazinthen, (E)-2-Hexenal einen blumigen Duft.[1]
Benzaldehyd wird in großen Mengen als Duftstoff sowie als Aroma in Speisen und Getränken eingesetzt, im Bereich Aromastoffe ist es eine der meistgenutzten Verbindungen.[203] Aldehyde spielen eine zentrale Rolle unter den Aromastoffen für Lebensmittel; um 1998 waren die meistgenutzten Aromastoffe Vanillin und Benzaldehyd.[204]
Sonstige Verwendung
[Bearbeiten | Quelltext bearbeiten]Formaldehyd wird weit überwiegend in der Herstellung von anderen Chemikalien und Kunststoffen verwendet (siehe oben). Die wichtigste Anwendung ohne Weiterverarbeitung ist als Desinfektions- und Konservierungsmittel, beispielsweise im medizinischen Bereich.[192] In der Medizin werden Formaldehyd und Glutaraldehyd als Flächen- und Instrumentendesinfektionsmittel eingesetzt. Beide Aldehyde haben eine gute Wirksamkeit gegen viele verschiedene Mikroorganismen. Insbesondere unbehüllte Viren und sporenbildende Bakterien (z. B. Milzbrand), die nur wenigen Desinfektionsmitteln zugänglich sind, können so erreicht werden. Da Aldehyde irritierend auf Haut und Schleimhäute wirken und gelegentlich Allergien auslösen, muss mit diesen Mitteln sorgfältig umgegangen werden.[205] Formaldehyd selbst wird als Konservierungsmittel in Kosmetika praktisch nicht mehr verwendet. Stattdessen kommen Formaldehyd-Abspalter, beispielsweise 2-Brom-2-nitropropan-1,3-diol, zum Einsatz, die länger wirken, aber nur eine geringe Konzentration an Formaldehyd erzeugen.[206] Eine standardmäßig als Fixierungsmittel verwendete Lösung ist 10%iges Formalin, was einer wässrigen Lösung mit 3,7 % Formaldehyd und 1 % Methanol entspricht.[207]
Nachweise
[Bearbeiten | Quelltext bearbeiten]Obwohl die NMR-Spektroskopie und anderer spektroskopischer Methoden chemische Nachweise funktioneller Gruppen zu Seltenheiten hat werden lassen, wird diese in Spezialfällen noch verwendet.[6] Aldehyde können mit der Tollensprobe, der Fehlingprobe und der Schiffsche Probe nachgewiesen werden.[208] Der Nachweis per Tollens- und Fehlingprobe beruht darauf, dass sie (im Gegensatz zu Ketonen) schon mit schwachen Oxidationsmitteln zu Carbonsäuren oxidieren.[15] Aromatische Aldehyde sind sich in ihrem chemischen Verhalten sehr ähnlich, sodass die Bestimmung einzelner Aldehyde mit den klassischen analytischen Methoden schwierig ist, wenn mehrere in der Probe vorhanden sind.[209]
Weblinks
[Bearbeiten | Quelltext bearbeiten]{kind=link}
{kind=link}
Wiktionary: Aldehyd – Bedeutungserklärungen, Wortherkunft, Synonyme, Übersetzungen
Einzelnachweise
[Bearbeiten | Quelltext bearbeiten]- ↑ a b c d e f Fritz Ullmann: Ullmann's encyclopedia of industrial chemistry. 5th ed Auflage. Band1. VCH, Weinheim Deerfield beach (Fla.) 1985, ISBN 3-527-20101-7 (englisch).
- ↑ Ira Remsen: Justus von Liebig, His Life and Work (1803–1873). By W. A. Shenstone, F. I. C., Lecturer on Chemistry in Clifton College. New York, Macmillan & Co. 1895. Pp. 220 + vi. In: Science. Band3, Nr.53, 3. Januar 1896, S.62–63, doi:10.1126/science.3.53.24 (englisch).
- ↑ Holde Puchtler, Susan N. Meloan, Barbara R. Brewton: On the history of basic fuchsin and aldehyde-schiff reactions from 1862 to 1935. In: Histochemistry. Band41, Nr.3, 1975, S.185–194, doi:10.1007/BF00497682 (englisch).
- ↑ Chérifa Boulechfar, Hana Ferkous, Amel Delimi, Amel Djedouani, Abdesalem Kahlouche, Abir Boublia, Ahmad S. Darwish, Tarek Lemaoui, Rajesh Verma, Yacine Benguerba: Schiff bases and their metal Complexes: A review on the history, synthesis, and applications. In: Inorganic Chemistry Communications. Band150, April 2023, S.110451, doi:10.1016/j.inoche.2023.110451 (englisch).
- ↑ Willi Melber, Peter Böhm: Einführung in die Nomenklatur organisch-chemischer Verbindungen für Studium und Berufsausbildung. Springer Berlin Heidelberg, 1987, ISBN 3-642-93370-X, S.75 (eingeschränkte Vorschau in der Google-Buchsuche).
- ↑ a b c d Kurt Peter C. Vollhardt, Neil Eric Schore: Organische Chemie. Haupbd. 6. Auflage. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 2020, ISBN 978-3-527-34582-3.
- ↑ S. 910
- ↑ Karl-Heinz Hellwich: Chemische Nomenklatur: die systematische Benennung organisch-chemischer Verbindungen; ein Lehrbuch für Pharmazie- und Chemiestudenten. 3., überarb. Auflage. Govi-Verl, Eschborn 1998, ISBN 3-7741-1095-6.
- ↑ Karl-Heinz Hellwich: Chemische Nomenklatur: die systematische Benennung organisch-chemischer Verbindungen; ein Lehrbuch für Pharmazie- und Chemiestudenten. 3., überarb. Auflage. Govi-Verl, Eschborn 1998, ISBN 3-7741-1095-6, S.94.
- ↑ Richard Cammack: Oxford dictionary of biochemistry and molecular biology. Second revised ed Auflage. Oxford university press, Oxford 2006, ISBN 0-19-852917-1 (englisch).
- ↑ The IUPAC Compendium of Chemical Terminology: The Gold Book. 4. Auflage. International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, NC 2019, doi:10.1351/goldbook.a00208 (englisch).
- ↑ The IUPAC Compendium of Chemical Terminology: The Gold Book. 4. Auflage. International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, NC 2019, doi:10.1351/goldbook.k03386 (englisch).
- ↑ Ameisensäure. In: Spektrum.de Lexikon der Chemie. Abgerufen am 5. Juni 2024.
- ↑ Paula Y. Bruice: Organische Chemie - Studieren kompakt (= Always learning). 5., aktualisierte Auflage. Pearson Studium, München 2011, ISBN 978-3-86894-102-9, S.673.
- ↑ Paula Y. Bruice: Organische Chemie - Studieren kompakt (= Always learning). 5., aktualisierte Auflage. Pearson Studium, München 2011, ISBN 978-3-86894-102-9, S.711.
- ↑ a b Adalbert Wollrab: Organische Chemie Eine Einführung für Lehramts- und Nebenfachstudenten. Springer-Verlag, 2013, ISBN 978-3-662-09137-1, S.402ff. (eingeschränkte Vorschau in der Google-Buchsuche).
- ↑ Quade R. Stahl: Preliminary Air Pollution Survey of Aldehydes: A Literature Review. U.S. Department of Health, Education, and Welfare, Public Health Service, Consumer Protection and Environmental Health Service, National Air Pollution Control Administration, 1969 (google.de [abgerufen am 28. März 2026]).
- ↑ George Socrates: Infrared and Raman Characteristic Group Frequencies. Wiley, 2004, ISBN 0-470-09307-2, S.124 (englisch, eingeschränkte Vorschau in der Google-Buchsuche).
- ↑ David R. Lide (Hrsg.): CRC Handbook of Chemistry and Physics. 90. Auflage. (Internet-Version: 2010), CRC Press / Taylor and Francis, Boca Raton FL, Physical Constants of Organic Compounds, S. 3-1 – 3-523.
- ↑ Siegfried Hauptmann: Organische Chemie. 2. durchgesehene Auflage, VEB Deutscher Verlag für Grundstoffindustrie, Leipzig, 1985, S. 565, ISBN 3-342-00280-8.
- ↑ Mikko Salaspuro: Acetaldehyde and gastric cancer. In: Journal of Digestive Diseases. Band12, Nr.2, April 2011, S.51–59, doi:10.1111/j.1751-2980.2011.00480.x (englisch).
- ↑ David Stewart: The Chemistry of Essential Oils Made Simple: God's Love Manifest in Molecules. Care Publications, 2005, ISBN 0-934426-99-6 (S. 324 in der Google-Buchsuche).
- ↑ Gulzar Ahmad Nayik, Amir Gull, Tariq Ahmad Ganaie: Handbook of Oleoresins: Extraction, Characterization, and Applications. CRC Press, 2022, ISBN 978-1-00-059266-5, S.68−69 (S. 68 in der Google-Buchsuche).
- ↑ Famous Erhunmwunsee, Chao Pan, Kunlong Yang, Yongxin Li, Man Liu, Jun Tian: Recent development in biological activities and safety concerns of perillaldehyde from perilla plants: A review. In: Critical Reviews in Food Science and Nutrition. Band62, Nr.23, 11. August 2022, S.6328–6340, doi:10.1080/10408398.2021.1900060.
- ↑ Eberhard Breitmaier: Terpene: Aromen, Düfte, Pharmaka, Pheromone. 1. Auflage. Wiley, 2005, ISBN 978-3-527-31498-0, doi:10.1002/9783527623693 (wiley.com [abgerufen am 29. März 2026]).
- ↑ Oliver P. Ernst, David T. Lodowski, Marcus Elstner, Peter Hegemann, Leonid S. Brown, Hideki Kandori: Microbial and Animal Rhodopsins: Structures, Functions, and Molecular Mechanisms. In: Chemical Reviews. Band114, Nr.1, 8. Januar 2014, S.126–163, doi:10.1021/cr4003769, PMID 24364740, PMC 3979449 (freier Volltext).
- ↑ NIIR Board of Consultants & Engineers: The Complete book on Natural Dyes & Pigments: How to Make Natural Purple Dyes From Plants, growing, harvesting and using natural dye plants, Making and Using Natural dyes plants, Dyeing Wool with Natural Plant Dyes, How to make plant based dyes,Natural dyes and dyeing from woodland plants. ASIA PACIFIC BUSINESS PRESS Inc., 2005, ISBN 978-81-7833-032-7, S.26, 30 (google.de [abgerufen am 29. März 2026]).
- ↑ Sylviane Liotenberg, Helen North, Annie Marion-Poll: Molecular biology and regulation of abscisic acid biosynthesis in plants. In: Plant Physiology and Biochemistry. Band37, Nr.5, Mai 1999, S.341–350, doi:10.1016/S0981-9428(99)80040-0 (elsevier.com [abgerufen am 29. März 2026]).
- ↑ Regulatory Mechanism in Vertebrates. Rastogi Publications, ISBN 978-81-7133-750-7 (google.de [abgerufen am 29. März 2026]).
- ↑ T. Swain: Chemical plant Taxonomy. Elsevier, 2012, ISBN 978-0-323-14624-1, S.350−353 (S. 350 in der Google-Buchsuche).
- ↑ Tiwatt Kuljanabhagavad, Piyanut Thongphasuk, Walee Chamulitrat, Michael Wink: Triterpene saponins from Chenopodium quinoa Willd. In: Phytochemistry. Band69, Nr.9, Juni 2008, S.1919–1926, doi:10.1016/j.phytochem.2008.03.001.
- ↑ Lutz‐F. Tietze: Secologanin, a Biogenetic Key Compound—Synthesis and Biogenesis of the Iridoid and Secoiridoid Glycosides. In: Angewandte Chemie International Edition in English. Band22, Nr.11, November 1983, S.828–841, doi:10.1002/anie.198308281.
- ↑ Lucy I. Darakjian, Aimilia Rigakou, Andrew Brannen, Mohammed H. Qusa, Niki Tasiakou, Panagiotis Diamantakos, Miranda N. Reed, Peter Panizzi, Melissa D. Boersma, Eleni Melliou, Khalid A. El Sayed, Prokopios Magiatis, Amal Kaddoumi: Spontaneous In Vitro and In Vivo Interaction of (−)-Oleocanthal with Glycine in Biological Fluids: Novel Pharmacokinetic Markers. In: ACS Pharmacology & Translational Science. Band4, Nr.1, 12. Februar 2021, ISSN 2575-9108, S.179–192, doi:10.1021/acsptsci.0c00166, PMID 33615171, PMC 7887843 (freier Volltext) – (acs.org [abgerufen am 26. März 2026]).
- ↑ Nobuhiro Shimizu, Ryota Yakumaru, Tomoyo Sakata, Satoshi Shimano, Yasumasa Kuwahara: The Absolute Configuration of Chrysomelidial: A Widely Distributed Defensive Component Among Oribotririid Mites (Acari: Oribatida). In: Journal of Chemical Ecology. Band38, Nr.1, Januar 2012, S.29–35, doi:10.1007/s10886-012-0064-3.
- ↑ M. Rowell-Rahier, J. M. Pasteels: Economics of chemical defense in chrysomelinae. In: Journal of Chemical Ecology. Band12, Nr.5, Mai 1986, S.1189–1203, doi:10.1007/BF01639004.
- ↑ J. P. Felix D'Mello: Handbook of Plant and Fungal Toxicants. CRC Press, 1997, ISBN 0-8493-8551-2, S.87−88 (S. 87 in der Google-Buchsuche).
- ↑ Taotao Ling, Jing Xu, Ryan Smith, Abbas Ali, Charles L. Cantrell, Emmanuel A. Theodorakis: Synthesis of (−)-callicarpenal, a potent arthropod repellent. In: Tetrahedron. Band67, Nr.17, April 2011, S.3023–3029, doi:10.1016/j.tet.2011.02.078, PMID 21643472, PMC 3105892 (freier Volltext) – (elsevier.com [abgerufen am 26. März 2026]).
- ↑ Hermann Schildknecht: Chemical Ecology—A Chapter of Modern Natural Products Chemistry. In: Angewandte Chemie International Edition in English. Band15, Nr.4, April 1976, ISSN 0570-0833, S.214–222, doi:10.1002/anie.197602141 (wiley.com [abgerufen am 29. März 2026]).
- ↑ James R. Hanson: Chemistry of Fungi. Royal Society of Chemistry, 2008, ISBN 978-0-85404-136-7 (google.de [abgerufen am 29. März 2026]).
- ↑ Peter Spiteller: Chemical Defence Strategies of Higher Fungi. In: Chemistry – A European Journal. Band14, Nr.30, 20. Oktober 2008, ISSN 0947-6539, S.9100–9110, doi:10.1002/chem.200800292 (wiley.com [abgerufen am 29. März 2026]).
- ↑ H.-D. Belitz, Werner Grosch, Peter Schieberle: Food Chemistry. Springer Science & Business Media, 2009, ISBN 978-3-540-69933-0, S.248−250 (google.de [abgerufen am 26. März 2026]).
- ↑ Hans Grisebach, Rolf Schmid: Chemistry and Biochemistry of Branched‐Chain Sugars. In: Angewandte Chemie International Edition in English. Band11, Nr.3, März 1972, ISSN 0570-0833, S.159–173, doi:10.1002/anie.197201591 (wiley.com [abgerufen am 29. März 2026]).
- ↑ Hans G. Schlegel, C. Zaborosch: General Microbiology. Cambridge University Press, 1993, ISBN 978-0-521-43980-0 (google.de [abgerufen am 29. März 2026]).
- ↑ Brian L. Carlson, Edward R. Ballister, Emmanuel Skordalakes, David S. King, Mark A. Breidenbach, Sarah A. Gilmore, James M. Berger, Carolyn R. Bertozzi: Function and Structure of a Prokaryotic Formylglycine-generating Enzyme. In: Journal of Biological Chemistry. Band283, Nr.29, Juli 2008, S.20117–20125, doi:10.1074/jbc.M800217200 (elsevier.com [abgerufen am 26. März 2026]).
- ↑ David R. Eyre, Mercedes A. Paz, Paul M. Gallop: CROSS-LINKING IN COLLAGEN AND ELASTIN. In: Annual Review of Biochemistry. Band53, Nr.1, Juni 1984, S.717–748, doi:10.1146/annurev.bi.53.070184.003441 (englisch).
- ↑ Francesca Ghirga, Deborah Quaglio, Patrizio Ghirga, Simone Berardozzi, Giovanni Zappia, Bruno Botta, Mattia Mori, Ilaria D'Acquarica: Occurrence of Enantioselectivity in Nature: The Case of ( S )‐Norcoclaurine. In: Chirality. Band28, Nr.3, März 2016, S.169–180, doi:10.1002/chir.22566.
- ↑ Michael Wink: Quinolizidine and Pyrrolizidine Alkaloid Chemical Ecology – a Mini-Review on Their Similarities and Differences. In: Journal of Chemical Ecology. Band45, Nr.2, Februar 2019, ISSN 0098-0331, S.109–115, doi:10.1007/s10886-018-1005-6 (springer.com [abgerufen am 29. März 2026]).
- ↑ Eric David Morgan: Biosynthesis in Insects. Royal Society of Chemistry, 2010, ISBN 978-1-84755-808-4, S.319−320 (google.de [abgerufen am 28. März 2026]).
- ↑ Edoardo Sarubbi, Pier Fausto Seneci, Michael R. Angelastro, Norton P. Peet, Maurizio Denaro, Khalid Islam: Peptide aldehydes as inhibitors of HIV protease. In: FEBS Letters. Band319, Nr.3, 22. März 1993, ISSN 0014-5793, S.253–256, doi:10.1016/0014-5793(93)80557-B (wiley.com [abgerufen am 26. März 2026]).
- ↑ Johan Hoebeke, Catherine Busatto‐Samsoen, Daniel Davoust, Evelyne Lebrun: 1 H NMR study of the diastereomeric forms of the protease inhibitor antipain. In: Magnetic Resonance in Chemistry. Band32, Nr.4, April 1994, ISSN 0749-1581, S.220–224, doi:10.1002/mrc.1260320406 (wiley.com [abgerufen am 26. März 2026]).
- ↑ Peter R. Cheeke: Toxicants of Plant Origin: Glycosides. CRC Press, 1989, ISBN 0-8493-6991-6, S.50 (google.de [abgerufen am 26. März 2026]).
- ↑ Raquel Sánchez-Pérez, Elizabeth HJ. Neilson: The case for sporadic cyanogenic glycoside evolution in plants. In: Current Opinion in Plant Biology. Band81, Oktober 2024, S.102608, doi:10.1016/j.pbi.2024.102608 (elsevier.com [abgerufen am 29. März 2026]).
- ↑ Eran Pichersky, Natalia Dudareva: Biology of Plant Volatiles. CRC Press, 2020, ISBN 978-0-429-84925-1, 8.2.2 Biosynthesis of Volatile Phenylpropanoids and Related Compounds (google.de [abgerufen am 29. März 2026]).
- ↑ a b István Ujváry, Joseph C. Dickens, James A. Kamm, Leslie M. McDonough: Natural product analogs: Stable mimics of aldehyde pheromones. In: Archives of Insect Biochemistry and Physiology. Band22, Nr.3-4, Januar 1993, S.393–411, doi:10.1002/arch.940220308 (englisch).
- ↑ Russell Jurenka: Fatty Acid Origin of Insect Pheromones. In: Insect Lipid Metabolism. Band1494. Springer Nature Switzerland, Cham 2024, ISBN 978-3-03204841-7, S.501–518, doi:10.1007/978-3-032-04842-4_813.
- ↑ T.L. Payne, W.E. Finn: Pheromone receptor system in the females of the greater wax moth Galleria mellonella. In: Journal of Insect Physiology. Band23, Nr.7, Januar 1977, S.879–881, doi:10.1016/0022-1910(77)90014-2 (englisch).
- ↑ a b Kento Yoshimori, Chika Okuda, Shinji Ohta, Hisashi Ômura: Sex pheromones from male forewings of the Common Grass Yellow Eurema mandarina. In: Journal of Chemical Ecology. Band48, Nr.5-6, Juni 2022, S.518–530, doi:10.1007/s10886-022-01368-0 (englisch).
- ↑ Eva Mishor, Daniel Amir, Tali Weiss, Danielle Honigstein, Aharon Weissbrod, Ethan Livne, Lior Gorodisky, Shiri Karagach, Aharon Ravia, Kobi Snitz, Diyala Karawani, Rotem Zirler, Reut Weissgross, Timna Soroka, Yaara Endevelt-Shapira, Shani Agron, Liron Rozenkrantz, Netta Reshef, Edna Furman-Haran, Heinz Breer, Joerg Strotmann, Tatsuya Uebi, Mamiko Ozaki, Noam Sobel: Sniffing the human body volatile hexadecanal blocks aggression in men but triggers aggression in women. In: Science Advances. Band7, Nr.47, 19. November 2021, doi:10.1126/sciadv.abg1530, PMID 34797713, PMC 8604408 (freier Volltext) – (englisch).
- ↑ Claus Wasternack: Oxylipins: Biosynthesis, Signal Transduction and Action. In: Annual Plant Reviews Volume 24: Plant Hormone Signaling. 1. Auflage. Wiley, 2006, ISBN 1-4051-3887-4, S.189–192, doi:10.1002/9780470988800.ch7 (englisch).
- ↑ Kenji Matsui: Green leaf volatiles: hydroperoxide lyase pathway of oxylipin metabolism. In: Current Opinion in Plant Biology. Band9, Nr.3, Juni 2006, S.274–280, doi:10.1016/j.pbi.2006.03.002 (englisch).
- ↑ Yuying Tang, Yijun Yao, Yangyun Wu, Shunbo Yang: The Volatile Composition, Biosynthesis Pathways, Breeding Strategies, and Regulation Measures of Apple Aroma: A Review. In: Horticulturae. Band11, Nr.3, 12. März 2025, S.310, doi:10.3390/horticulturae11030310 (englisch).
- ↑ Chenxi Ji, Chenxu Liu, Rongqing Wang, Meiying Ruan, Zhuping Yao, Hongjian Wan, Zhimiao Li, Yuan Cheng, Qingjing Ye: The Science behind vegetable aromas: types, synthesis, and influencing factors of volatile compounds. In: Plant Signaling & Behavior. Band20, Nr.1, 30. April 2025, doi:10.1080/15592324.2025.2527958, PMID 40650942, PMC 12258172 (freier Volltext) – (englisch).
- ↑ Agata Jabłońska‐Trypuć, Walentyn Pankiewicz, Romuald Czerpak: Traumatic Acid Reduces Oxidative Stress and Enhances Collagen Biosynthesis in Cultured Human Skin Fibroblasts. In: Lipids. Band51, Nr.9, September 2016, S.1021–1035, doi:10.1007/s11745-016-4174-5, PMID 27423205, PMC 5009161 (freier Volltext) – (englisch).
- ↑ Angelo Fontana, Giuliana d'Ippolito, Adele Cutignano, Antonio Miralto, Adrianna Ianora, Giovanna Romano, Guido Cimino: Chemistry of oxylipin pathways in marine diatoms. In: Pure and Applied Chemistry. Band79, Nr.4, 1. Januar 2007, ISSN 1365-3075, S.481–490, doi:10.1351/pac200779040481 (degruyter.com [abgerufen am 29. März 2026]).
- ↑ Ian Tizard, Loren Skow: The olfactory system: the remote-sensing arm of the immune system. In: Animal Health Research Reviews. Band22, Nr.1, Juni 2021, ISSN 1466-2523, S.14–25, doi:10.1017/S1466252320000262 (cambridge.org [abgerufen am 29. März 2026]).
- ↑ Klaus Urich: Comparative Animal Biochemistry. Springer Science & Business Media, 1994, ISBN 978-3-540-57420-0 (google.de [abgerufen am 29. März 2026]).
- ↑ E A Meighen: Molecular biology of bacterial bioluminescence. In: Microbiological Reviews. Band55, Nr.1, März 1991, S.124−125, doi:10.1128/mr.55.1.123-142.1991, PMID 2030669, PMC 372803 (freier Volltext).
- ↑ Pergamon titles of related interest. In: Enzymes Dependent on Pyridoxal Phosphate and Other Carbonyl Compounds As Cofactors. Elsevier, 1991, ISBN 0-08-040820-6, S.1 (englisch).
- ↑ Zhehao Jin, Dae-Kyun Ro, Soo-Un Kim, Moonhyuk Kwon: Piperonal synthase from black pepper (Piper nigrum) synthesizes a phenolic aroma compound, piperonal, as a CoA-independent catalysis. In: Applied Biological Chemistry. Band65, Nr.1, Dezember 2022, ISSN 2468-0834, doi:10.1186/s13765-022-00691-0, PMID 35402752, PMC 8948145 (freier Volltext) – (springeropen.com [abgerufen am 26. März 2026]).
- ↑ Anish Kundu: Vanillin biosynthetic pathways in plants. In: Planta. Band245, Nr.6, Juni 2017, ISSN 0032-0935, S.1069–1078, doi:10.1007/s00425-017-2684-x (springer.com [abgerufen am 26. März 2026]).
- ↑ Howell G.M. Edwards, Emma M. Newton, David D. Wynn-Williams: Molecular structural studies of lichen substances II: atranorin, gyrophoric acid, fumarprotocetraric acid, rhizocarpic acid, calycin, pulvinic dilactone and usnic acid. In: Journal of Molecular Structure. Band651-653, Juni 2003, S.27–37, doi:10.1016/S0022-2860(02)00626-9 (elsevier.com [abgerufen am 26. März 2026]).
- ↑ Dagmar Kock, Joachim Ruther, Klaus Peter Sauer: A Male Sex Pheromone in a Scorpionfly. In: Journal of Chemical Ecology. Band33, Nr.6, Juni 2007, S.1249–1256, doi:10.1007/s10886-007-9304-3 (englisch).
- ↑ Kevin R. Ulrich, Matthew Kramer, Mark F. Feldlaufer: Ability of bed bug ( H emiptera: C imicidae) defensive secretions ( E )‐2‐hexenal and ( E )‐2‐octenal to attract adults of the common bed bug C imex lectularius. In: Physiological Entomology. Band41, Nr.2, Juni 2016, S.103–110, doi:10.1111/phen.12129 (englisch).
- ↑ Joanna Domagalska, Anna Janas, Krystian Pyta, Paulina Pecyna, Piotr Ruszkowski, Lech Celewicz, Marzena Gajecka, Franz Bartl, Piotr Przybylski: 16‐Membered Macrolide Lactone Derivatives Bearing a Triazole‐Functionalized Arm at the Aglycone C13 Position as Antibacterial and Anticancer Agents. In: ChemMedChem. Band11, Nr.17, 6. September 2016, S.1886–1891, doi:10.1002/cmdc.201600250.
- ↑ Herbert A. Kirst, John E. Toth, Manuel Debono, Kevin E. Willard, Brenda A. Truedell, John L. Ott, Fred T. Counter, Anna M. Felty-Duckworth, Robert S. Pekarek: Synthesis and evaluation of tylosin-related macrolides modified at the aldehyde function: a new series of orally effective antibiotics. In: Journal of Medicinal Chemistry. Band31, Nr.8, August 1988, S.1631–1641, doi:10.1021/jm00403a025.
- ↑ Ludmila Chistoserdova: Modularity of methylotrophy, revisited. In: Environmental Microbiology. Band13, Nr.10, Oktober 2011, ISSN 1462-2912, S.2603–2622, doi:10.1111/j.1462-2920.2011.02464.x (wiley.com [abgerufen am 29. März 2026]).
- ↑ B. Zuckerman, David Buhl, Patrick Palmer, Lewis E. Snyder: Observations of Interstellar Formaldehyde. In: The Astrophysical Journal. Band160, Mai 1970, ISSN 0004-637X, S.485, doi:10.1086/150449.
- ↑ Jia Wang, Chaojiang Zhang, Joshua H. Marks, Mikhail M. Evseev, Oleg V. Kuznetsov, Ivan O. Antonov, Ralf I. Kaiser: Interstellar formation of lactaldehyde, a key intermediate in the methylglyoxal pathway. In: Nature Communications. Band15, Nr.1, 24. November 2024, ISSN 2041-1723, doi:10.1038/s41467-024-54562-x, PMID 39582038, PMC 11586434 (freier Volltext) – (nature.com [abgerufen am 28. März 2026]).
- ↑ H. James Cleaves II: The prebiotic geochemistry of formaldehyde. In: Precambrian Research. Band164, Nr.3-4, 30. Juli 2008, S.111–118, doi:10.1016/j.precamres.2008.04.002 (elsevier.com [abgerufen am 28. März 2026]).
- ↑ Arthur L. Weber: Chemical Constraints Governing the Origin of Metabolism: The Thermodynamic Landscape of Carbon Group Transformations under Mild Aqueous Conditions. In: Origins of life and evolution of the biosphere. Band32, Nr.4, August 2002, ISSN 0169-6149, S.333–357, doi:10.1023/A:1020588925703 (springer.com [abgerufen am 28. März 2026]).
- ↑ Fude Chen, Yufen Zhao, Jianxi Ying: From searching for extraterrestrial biosignatures to exploring the origins of life: A prebiotic chemistry perspective. In: Fundamental Research. Februar 2026, doi:10.1016/j.fmre.2026.01.017 (elsevier.com [abgerufen am 28. März 2026]).
- ↑ Nicholas J. Lawrence: Aldehydes and ketones. In: Journal of the Chemical Society, Perkin Transactions 1. Nr.10, 1998, S.1739–1750, doi:10.1039/a800646f (englisch).
- ↑ Gabriel Tojo, Marcos Fernandez: Oxidation of alcohols to aldehydes and ketones: a guide to current common practice (= Basic reactions in organic synthesis). Springer, New York, NY 2006, ISBN 0-387-23607-4 (englisch).
- ↑ Francis A. Carey, Richard J. Sundberg: Advanced Organic Chemistry: Part B: Reaction and Synthesis. Springer Science & Business Media, 2007, ISBN 978-0-387-68350-8 (englisch).
- ↑ Kurt Peter C. Vollhardt, Neil Eric Schore: Organische Chemie. John Wiley & Sons, 2011, ISBN 978-3-527-32754-6.
- ↑ Francis A. Carey, Richard J. Sundberg: Advanced Organic Chemistry: Part B: Reaction and Synthesis. Springer, 2006, ISBN 0-306-47380-1 (englisch).
- ↑ Laszlo Kurti, Barbara Czako: Strategic Applications of Named Reactions in Organic Synthesis. Elsevier, 2005, ISBN 0-08-057541-2 (englisch).
- ↑
Comprehensive Organic Synthesis. Newnes, 2014, ISBN 978-0-08-097743-0 (englisch).
- ↑ Volume 1, S. 751 in der Google-Buchsuche
- ↑ Volume 1, S. 750 in der Google-Buchsuche
- ↑ Volume 1, S. 749 in der Google-Buchsuche
- ↑ Volume 8, S. 437-438 in der Google-Buchsuche
- ↑ Jie Jack Li, E. J. Corey: Name Reactions of Functional Group Transformations. John Wiley & Sons, 2007, ISBN 978-0-470-17650-4 (englisch).
- ↑ Adalbert Wollrab: Organische Chemie: Eine Einführung für Lehramts- und Nebenfachstudenten. Springer-Verlag, 2013, ISBN 978-3-662-09137-1.
- ↑ Jie Jack Li: Name Reactions: A Collection of Detailed Reaction Mechanisms. Springer Science & Business Media, 2013, ISBN 978-3-662-05336-2 (englisch).
- ↑ P. Henry: Palladium Catalyzed Oxidation of Hydrocarbons. Springer Science & Business Media, 1979, ISBN 90-277-0986-6, S.41–46 (englisch, S. 41-46 in der Google-Buchsuche).
- ↑ Xiao-Feng Wu: Solvents as Reagents in Organic Synthesis: Reactions and Applications. John Wiley & Sons, 2018, ISBN 978-3-527-34196-2, S.9–10 (englisch, S. 9-10 in der Google-Buchsuche).
- ↑
Reinhard Brückner: Organic Mechanisms: Reactions, Stereochemistry and Synthesis. Springer Science & Business Media, 2010, ISBN 978-3-642-03650-7 (englisch).
- ↑ S. 766-769 in der Google-Buchsuche
- ↑ S. 769-770 in der Google-Buchsuche
- ↑ Helmut Krauch, Werner Kunz: Reaktionen der organischen Chemie. John Wiley & Sons, 2009, ISBN 978-3-527-62512-3.
- ↑ Sivaraman Balasubramaniam, Indrapal Aidhen: The Growing Synthetic Utility of the Weinreb Amide. In: Synthesis. Band2008, Nr.23, Dezember 2008, S.3707–3738, doi:10.1055/s-0028-1083226 (englisch).
- ↑ Jonathan M. White, Ashok Rao Tunoori, Gunda I. Georg: A Novel and Expeditious Reduction of Tertiary Amides to Aldehydes Using Cp 2 Zr(H)Cl. In: Journal of the American Chemical Society. Band122, Nr.48, 1. Dezember 2000, S.11995–11996, doi:10.1021/ja002149g (englisch).
- ↑ Thomas Laue, Andreas Plagens: Named Organic Reactions. John Wiley & Sons, 2005, ISBN 0-470-02644-8 (englisch).
- ↑ Jie Jack Li: Name Reactions: A Collection of Detailed Reaction Mechanisms. Springer Science & Business Media, 2003, ISBN 3-540-40203-9 (englisch).
- ↑ S. Chandrasekhar, M. Suresh Kumar, B. Muralidhar: One pot conversion of carboxylic acids to aldehydes with DIBAL-H. In: Tetrahedron Letters. Band39, Nr.8, Februar 1998, S.909–910, doi:10.1016/S0040-4039(97)10688-8 (englisch).
- ↑ Jie Jack Li: Name Reactions: A Collection of Detailed Reaction Mechanisms. Springer Science & Business Media, 2003, ISBN 3-540-40203-9 (englisch).
- ↑ Xiao-Feng Wu, Buxing Han, Kuiling Ding, Zhongmin Liu: The Chemical Transformations of C1 Compounds. John Wiley & Sons, 2022, ISBN 978-3-527-83189-0, S.325–327 (englisch, S. 325-327 in der Google-Buchsuche).
- ↑ Bradford P. Mundy, Michael G. Ellerd, Frank G. Favaloro Jr: Name Reactions and Reagents in Organic Synthesis. John Wiley & Sons, 2005, ISBN 0-471-73986-3, S.162 (englisch, S. 162 in der Google-Buchsuche).
- ↑ Alexander R. Surrey: Name Reactions in Organic Chemistry. Elsevier, 2013, ISBN 978-1-4832-5868-3 (englisch).
- ↑ Shun-Ichi Murahashi: Ruthenium in Organic Synthesis. John Wiley & Sons, 2006, ISBN 3-527-60579-7, S.310–312 (englisch, S. 310-312 in der Google-Buchsuche).
- ↑ Alfred Hassner, Irishi Namboothiri, Meir Golan: Organic Syntheses Based on Name Reactions: A Practical Encyclopedic Guide to Nearly 800 Transformations. Elsevier, 2025, ISBN 978-0-08-102962-6 (englisch).
- ↑ Eberhard Breitmaier, Günther Jung: Organische Chemie. Thieme, 2005, ISBN 3-13-541505-8, S.319 (eingeschränkte Vorschau in der Google-Buchsuche).
- ↑ Hans P. Latscha, Helmut A. Klein: Organische Chemie. Springer Berlin Heidelberg, 2013, S.203 (eingeschränkte Vorschau in der Google-Buchsuche).
- ↑ Reinhard Bruckner: Advanced Organic Chemistry: Reaction Mechanisms. Elsevier, 2001, ISBN 0-08-049880-9, S.286–287 (englisch, S. 286-287 in der Google-Buchsuche).
- ↑
Hans-Georg Elias: Macromolecules: Volume 1 · Structure and Properties / Volume 2 · Synthesis and Materials. Springer Science & Business Media, 2013, ISBN 978-1-4615-7364-7 (englisch).
- ↑ S. 933-934 in der Google-Buchsuche
- ↑ S. 938-939 in der Google-Buchsuche
- ↑ H. Panda: Handbook On Chemical Industries (Alcohol Based). ASIA PACIFIC BUSINESS PRESS Inc., 2002, ISBN 81-7833-067-9, S.28–29 (englisch, S. 28-29 in der Google-Buchsuche).
- ↑ a b c Joachim Buddrus: Grundlagen der organischen Chemie. De Gruyter, 2020, ISBN 978-3-11-158631-1, S.452 (452 in der Google-Buchsuche).
- ↑ Michael B. Smith: March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure. John Wiley & Sons, 2025, ISBN 978-1-394-24299-3, S.875 (englisch, 875 in der Google-Buchsuche).
- ↑ Peter G. M. Wuts, Theodora W. Greene: Greene's Protective Groups in Organic Synthesis. 1. Auflage. Wiley, 2006, ISBN 0-471-69754-0, doi:10.1002/9780470053485.ch4 (englisch, Online [abgerufen am 6. März 2026]).
- ↑ Sabine Bognar, Manuel van Gemmeren: Direct Synthesis of Unsymmetrical Dithioacetals. In: Chemistry – A European Journal. Band27, Nr.15, 12. März 2021, S.4859–4863, doi:10.1002/chem.202004835, PMID 33270274, PMC 8048688 (freier Volltext) – (englisch).
- ↑
{{Literatur |Autor=Eberhard Breitmaier, Günther Jung |Titel=Organische Chemie: Grundlagen, Stoffklassen, Reaktionen, Konzepte, Molekülstruktur; zahlreiche Formeln, Tabellen |Auflage=5., überarb. Aufl |Verlag=Thieme |Ort=Stuttgart |Datum=2005 |ISBN=3-13-541505-8 |Sprache=de}
- ↑ S. 321 in der Google-Buchsuche
- ↑ S. 627-628 in der Google-Buchsuche
- ↑ James Keeler, Peter Wothers: Chemical Structure and Reactivity: An Integrated Approach. OUP Oxford, 2013, ISBN 978-0-19-960413-5 (S. 365 in der Google-Buchsuche).
- ↑ S. 365-368
- ↑ Comprehensive Organic Synthesis. Newnes, 2014, ISBN 978-0-08-097743-0 (englisch, Online [abgerufen am 5. März 2026]).
- ↑ Volume 8, S. 96-97 in der Google-Buchsuche
- ↑ Volume 8, S. 4
- ↑ Francis A. Carey, Richard J. Sundberg: Advanced organic chemistry. B: Reactions and synthesis. Corr. 2. print Auflage. Springer, New York 2008, ISBN 978-0-387-68354-6 (englisch).
- ↑ Leo A. Paquette: Chiral Reagents for Asymmetric Synthesis. John Wiley & Sons, 2003, ISBN 0-470-85625-4, S.32 (englisch, S. 32 in der Google-Buchsuche).
- ↑ Michael B. Smith: March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure. John Wiley & Sons, 2019, ISBN 978-1-119-37179-3, 16-34 The Aldol Condensation (englisch, Online [abgerufen am 7. März 2026]).
- ↑ Roberto Ballini, Alessandro Palmieri: Nitroalkanes: Synthesis, Reactivity, and Applications. John Wiley & Sons, 2021, ISBN 978-3-527-82676-6, S.72 (englisch, Online [abgerufen am 6. März 2026]).
- ↑ Jonathan Clayden, Nick Greeves, Stuart G. Warren: Organic chemistry. 2nd ed Auflage. Oxford University Press, Oxford; New YorK 2012, ISBN 978-0-19-927029-3, S.130–132 (englisch).
- ↑ Reinhard Brückner: Advanced organic chemistry: reaction mechanisms (= Advanced organic chemistry series). Harcourt/Academic Press, San Diego 2002, ISBN 0-12-138110-2, S.307–308 (englisch).
- ↑ Gordon W. Gribble, Ahmed F. Abdel-Magid: Sodium Triacetoxyborohydride. In: Encyclopedia of Reagents for Organic Synthesis. John Wiley & Sons, Ltd, Chichester, UK 2007, ISBN 978-0-471-93623-7 (englisch).
- ↑ Robert F. Nystrom, Weldon G. Brown: Reduction of Organic Compounds by Lithium Aluminum Hydride. I. Aldehydes, Ketones, Esters, Acid Chlorides and Acid Anhydrides. In: Journal of the American Chemical Society. Band69, Nr.5, Mai 1947, S.1197–1199, doi:10.1021/ja01197a060 (englisch).
- ↑ Mathias Glatz, Berthold Stöger, Daniel Himmelbauer, Luis F. Veiros, Karl Kirchner: Chemoselective Hydrogenation of Aldehydes under Mild, Base-Free Conditions: Manganese Outperforms Rhenium. In: ACS Catalysis. Band8, Nr.5, 4. Mai 2018, S.4009–4016, doi:10.1021/acscatal.8b00153, PMID 29755828, PMC 5939901 (freier Volltext) – (englisch).
- ↑ C. W. Jung, P. E. Garrou: Dehydrogenation of alcohols and hydrogenation of aldehydes using homogeneous ruthenium catalysts. In: Organometallics. Band1, Nr.4, April 1982, S.658–666, doi:10.1021/om00064a016 (englisch).
- ↑ A. L. Wilds: Reduction with Aluminum Alkoxides. In: Organic Reactions. 15. März 2011, S.179–180, doi:10.1002/0471264180.or002.05 (englisch).
- ↑ William H. Brown, Thomas Poon: Einführung in die Organische Chemie. Band 1. WILEY-VCH, Weinheim 2021, ISBN 978-3-527-34674-5, S.414.
- ↑ Ender Erdik: Organozinc reagents in organic synthesis (= New directions in organic and biological chemistry). CRC Press, Boca Raton 1996, ISBN 0-8493-9151-2, S.192 (englisch).
- ↑ Handbook of reagents for organic synthesis. Wiley, Chichester, West Sussex, UK; New York 1999, ISBN 0-471-97924-4, S.72 (englisch).
- ↑ Jie Jack Li: Name Reactions for Homologation, Part 1. John Wiley & Sons, 2009, ISBN 978-0-470-48701-3, S.299–300 (englisch, Online [abgerufen am 5. März 2026]).
- ↑ Timo Gehring, Mark Busch, Martin Schlageter, Daniel Weingand: A concise summary of experimental facts about the Soai reaction. In: Chirality. Band22, 1E, Januar 2010, doi:10.1002/chir.20849 (englisch, Online [abgerufen am 5. März 2026]).
- ↑ P. Tharmaraj, J. Shakina, R. Sujatha, L. Alphonse: Green Chemistry. AG PUBLISHING HOUSE (AGPH Books), ISBN 978-81-19-15262-9, S.113 (englisch, eingeschränkte Vorschau in der Google-Buchsuche).
- ↑ Yoshiharu Iwabuchi: Green Oxidative Synthesis of Aldehydes and Ketones. In: Green Oxidation in Organic Synthesis. 1. Auflage. Wiley, 2019, ISBN 978-1-119-30416-6, S.35–78, doi:10.1002/9781119304197.ch2 (englisch, Online [abgerufen am 3. März 2026]).
- ↑ S. 159-162
- ↑ Ajnish Kumar Gupta, Bharti Gupta: Mechanism of Organic Reactions: For World of competitions. Ajnish Kumar Gupta, 5. Mai 2020, S.113–114 (englisch, Online [abgerufen am 3. März 2026]).
- ↑ Stanley R. Sandler, Wolf Karo: Organic functional group preparations. In: Organic Chemistry - A Series of Monographs. 2nd ed Auflage. v. 12. Academic Press, New York 1983, ISBN 0-12-618601-4, S.305 (englisch).
- ↑ Elias J. Corey, Norman W. Gilman, B. E. Ganem: New methods for the oxidation of aldehydes to carboxylic acids and esters. In: Journal of the American Chemical Society. Band90, Nr.20, September 1968, S.5616–5617, doi:10.1021/ja01022a059 (englisch).
- ↑ Cheves. Walling, Michael J. Mintz: Positive Halogen Compounds. XIII. t-Butyl Hypochlorite Chlorination of Ethers, Aldehydes, and Other Molecules with Polar Substituents. In: Journal of the American Chemical Society. Band89, Nr.6, März 1967, S.1515–1519, doi:10.1021/ja00982a040 (englisch).
- ↑ Nitin D. Arote, Krishnacharya G. Akamanchi: Direct conversion of aldehydes to acyl azides using tert-butyl hypochlorite. In: Tetrahedron Letters. Band48, Nr.32, August 2007, S.5661–5664, doi:10.1016/j.tetlet.2007.06.020 (englisch).
- ↑ Dong Ho Kang, Tae Young Joo, Warinthorn Chavasiri, Doo Ok Jang: Radical mediated-direct conversion of aldehydes into acid bromides. In: Tetrahedron Letters. Band48, Nr.2, Januar 2007, S.285–287, doi:10.1016/j.tetlet.2006.11.013 (englisch, Online [abgerufen am 3. März 2026]).
- ↑ Yumeng Liang, Zhengyu Zhao, Akihito Taya, Norio Shibata: Acyl Fluorides from Carboxylic Acids, Aldehydes, or Alcohols under Oxidative Fluorination. In: Organic Letters. Band23, Nr.3, 5. Februar 2021, S.847–852, doi:10.1021/acs.orglett.0c04087 (englisch, Online [abgerufen am 3. März 2026]).
- ↑ Liyuan Lan, Shuai Huang, Yongguo Liu, Baoguo Sun, Hongyu Tian: Preparation and odor characteristics of nitriles derived from aldehydes. In: Flavour and Fragrance Journal. Band35, Nr.4, Juli 2020, S.425–434, doi:10.1002/ffj.3581 (englisch).
- ↑ Dylan J. Quinn, Graham J. Haun, Gustavo Moura-Letts: Direct synthesis of nitriles from aldehydes with hydroxylamine-O-sulfonic acid in acidic water. In: Tetrahedron Letters. Band57, Nr.34, August 2016, S.3844–3847, doi:10.1016/j.tetlet.2016.07.047 (englisch).
- ↑ Antonella Leggio, Emilia Lucia Belsito, Sonia Gallo, Angelo Liguori: One-pot conversion of aldehydes to nitriles mediated by TiCl 4. In: Tetrahedron Letters. Band58, Nr.15, April 2017, S.1512–1514, doi:10.1016/j.tetlet.2017.03.007 (englisch).
- ↑ Dr Bhimraj Gawade, Mr I. G. Nannaware: Chemistry of Named Organic Reactions and Reagents. Xoffencerpublication, 2022, ISBN 978-93-9470707-8 (englisch, Online [abgerufen am 7. März 2026]).
- ↑ William Carruthers, Iain Coldham: Modern methods of organic synthesis. 4. ed., 4. printing with corrections. Cambridge University Press, Cambridge 2006, ISBN 0-521-77097-1, S.457 (englisch).
- ↑ Michael Smith, Jerry March: March's advanced organic chemistry: reactions, mechanisms, and structure. Eighth edition Auflage. John Wiley, Hoboken, NJ 2020, ISBN 978-1-119-37179-3, Eintrag 19-66 (englisch).
- ↑ Guangkuan Zhao, Ling‐Zhi Yuan, Mouad Alami, Olivier Provot: Desulfurization of Thioketals into Methylene and Methyl Derivatives: Nickel or not Nickel? In: ChemistrySelect. Band2, Nr.33, 21. November 2017, S.10951–10959, doi:10.1002/slct.201702370 (englisch).
- ↑ Bo Han, Chunping Ren, Min Jiang, Lipeng Wu: Titanium‐Catalyzed Exhaustive Reduction of Oxo‐Chemicals. In: Angewandte Chemie International Edition. Band61, Nr.46, 14. November 2022, doi:10.1002/anie.202209232 (englisch).
- ↑ S. Chandrasekhar, Ch. Raji Reddy, B. Nagendra Babu: Rapid Defunctionalization of Carbonyl Group to Methylene with Polymethylhydrosiloxane−B(C 6 F 5 ) 3. In: The Journal of Organic Chemistry. Band67, Nr.25, 1. Dezember 2002, S.9080–9082, doi:10.1021/jo0204045 (englisch, Online [abgerufen am 6. März 2026]).
- ↑ Christopher M. Beck, Scott E. Rathmill, You Jung Park, Junyi Chen, Robert H. Crabtree, Louise M. Liable-Sands, Arnold L. Rheingold: Aldehyde Decarbonylation Catalysis under Mild Conditions. In: Organometallics. Band18, Nr.25, 1. Dezember 1999, S.5311–5317, doi:10.1021/om9905106 (englisch).
- ↑ Manfred T. Reetz: Dichlorodimethyltitanium. In: Encyclopedia of Reagents for Organic Synthesis. John Wiley & Sons, Ltd, Chichester 2001, ISBN 0-471-93623-5, doi:10.1002/047084289x.rd392 (englisch, Online [abgerufen am 5. März 2026]).
- ↑ Volume Editors. In: Comprehensive Organic Functional Group Transformations. Band1. Elsevier, 1995, ISBN 0-08-044705-8, S.720–722 (englisch).
- ↑ Francis A. Carey, Richard J. Sundberg: Advanced organic chemistry. Reactions and synthesis. 4. ed., repr. Springer, New York 2006, ISBN 0-306-46244-3, S.111–112 (englisch).
- ↑ Reinhard Brückner, Michael Harmata: Organic mechanisms: reactions, stereochemistry and synthesis. Springer, Berlin 2010, ISBN 978-3-642-03650-7, S.467–469 (englisch).
- ↑ László Kürti, Barbara Czakó: Strategic applications of named reactions in organic synthesis: background and detailed mechanisms; 250 named reactions. Nachdr. Auflage. Elsevier Acad. Press, Amsterdam Heidelberg 2009, ISBN 978-0-12-369483-6, S.212 (englisch).
- ↑ Comprehensive organic synthesis. Second edition Auflage. Band1. Elsevier, Amsterdam, Netherlands; Waltham, MA 2014, ISBN 978-0-08-099964-7, S.518–520 (englisch).
- ↑ Comprehensive organic synthesis. Second edition Auflage. Band1. Elsevier, Amsterdam, Netherlands; Waltham, MA 2014, ISBN 978-0-08-099964-7, S.529 (englisch).
- ↑ Modern carbonyl olefination. Wiley-VCH, Weinheim 2004, ISBN 3-527-30634-X, S.18 (englisch).
- ↑ a b Ian Beadham, Jason Micklefield: Reagents for Carbonyl Methylenation in Organic Synthesis. In: Current Organic Synthesis. Band2, Nr.2, S.231–259, doi:10.2174/1570179053545396 (englisch).
- ↑ a b c Bernhard Breit: Dithioacetals as an Entry to Titanium-Alkylidene Chemistry: A New and Efficient Carbonyl Olefination. In: Angewandte Chemie International Edition. Band37, Nr.4, 2. März 1998, S.453–456, doi:10.1002/(SICI)1521-3773(19980302)37:4<453::AID-ANIE453>3.0.CO;2-M (englisch).
- ↑ Name reactions for homologations. Wiley, Hoboken 2009, ISBN 978-0-470-08507-3, S.268–269 (englisch).
- ↑ a b Damien Habrant, Vesa Rauhala, Ari M. P. Koskinen: Conversion of carbonyl compounds to alkynes: general overview and recent developments. In: Chemical Society Reviews. Band39, Nr.6, 2010, S.2007, doi:10.1039/b915418c (englisch).
- ↑ E.J. Corey, P.L. Fuchs: A synthetic method for formyl→ethynyl conversion (RCHO→RCCH or RCCR′). In: Tetrahedron Letters. Band13, Nr.36, 1972, S.3769–3772, doi:10.1016/S0040-4039(01)94157-7 (englisch).
- ↑ Frédéric Eymery, Bogdan Iorga, Philippe Savignac: The Usefulness of Phosphorus Compounds in Alkyne Synthesis. In: Synthesis. Band2000, Nr.02, 2000, S.185–213, doi:10.1055/s-2000-6241 (englisch).
- ↑ R. S. Sagitullin, G. P. Shkil', I. I. NosonovA, A. A. Ferber: Synthesis of pyridine bases by the chichibabin method (review). In: Chemistry of Heterocyclic Compounds. Band32, Nr.2, Februar 1996, S.127–140, doi:10.1007/BF01165434 (englisch).
- ↑ K. Peter C. Vollhardt, Neil E. Schore: Organische Chemie. John Wiley & Sons, 2020, ISBN 978-3-527-82112-9, S.1375 (Online [abgerufen am 2. März 2026]).
- ↑ Jie Jack Li: Namensreaktionen: Eine Sammlung von detaillierten Mechanismen und Anwendungen in der Synthese. Springer-Verlag, 2024, ISBN 978-3-03152850-7, S.218 (Online [abgerufen am 6. März 2026]).
- ↑ Jie Jack Li: Name Reactions in Heterocyclic Chemistry. John Wiley & Sons, 2004, ISBN 0-471-70414-8, S.488–489 (englisch, Online [abgerufen am 6. März 2026]).
- ↑ Jie Jack Li: Name Reactions in Heterocyclic Chemistry. John Wiley & Sons, 2004, ISBN 0-471-70414-8, S.168–169 (englisch, Online [abgerufen am 6. März 2026]).
- ↑ Jie Jack Li: Name Reactions in Heterocyclic Chemistry. John Wiley & Sons, 2004, ISBN 0-471-70414-8, S.207–210 (englisch, Online [abgerufen am 6. März 2026]).
- ↑ Jie Jack Li: Name Reactions in Heterocyclic Chemistry. John Wiley & Sons, 2004, ISBN 0-471-70414-8, S.79 (englisch, Online [abgerufen am 6. März 2026]).
- ↑ M. R. Grimmett: Imidazole and Benzimidazole Synthesis. Academic Press, 1997, ISBN 0-08-053445-7, S.41 (englisch, Online [abgerufen am 6. März 2026]).
- ↑ Surya K. De: Applied Organic Chemistry: Reaction Mechanisms and Experimental Procedures in Medicinal Chemistry. John Wiley & Sons, 2021, ISBN 978-3-527-34785-8, S.378 (englisch, Online [abgerufen am 6. März 2026]).
- ↑ Jie Jack Li: Name Reactions in Heterocyclic Chemistry. John Wiley & Sons, 2004, ISBN 0-471-70414-8, S.2 (englisch, Online [abgerufen am 6. März 2026]).
- ↑ Masahito. Segi, Tadashi. Nakajima, Sohei. Suga, Shinji. Murai, Ilhyong. Ryu, Akiya. Ogawa, Noboru. Sonoda: Direct conversion of aldehydes to seleno- and thioaldehydes. In: Journal of the American Chemical Society. Band110, Nr.6, März 1988, S.1976–1978, doi:10.1021/ja00214a058 (englisch, Online [abgerufen am 5. März 2026]).
- ↑ Masahito Segi, Tadashi Koyama, Yukihiro Takata, Tadashi Nakajima, Sohei Suga: Telluro aldehydes and telluro ketones. In: Journal of the American Chemical Society. Band111, Nr.23, November 1989, S.8749–8751, doi:10.1021/ja00205a044 (englisch, Online [abgerufen am 5. März 2026]).
- ↑ Wilfred L. F. Armarego, Christina Li Lin Chai: Purification of laboratory chemicals. 6. ed Auflage. Butterworth-Heinemann, Amsterdam Heidelberg 2009, ISBN 978-1-85617-567-8, S.65 (englisch).
- ↑ Walter M. Lauer, Carl M. Langkammerer: The Constitution of the Bisulfite Addition Compounds of Aldehydes and Ketones. In: Journal of the American Chemical Society. Band57, Nr.12, Dezember 1935, S.2360–2362, doi:10.1021/ja01315a007 (englisch).
- ↑ Damian E. Yerien, Sebastian Barata‐Vallejo, Al Postigo: Difluoromethylation Reactions of Organic Compounds. In: Chemistry – A European Journal. Band23, Nr.59, 20. Oktober 2017, S.14676–14701, doi:10.1002/chem.201702311 (englisch, Online [abgerufen am 6. März 2026]).
- ↑ Ulrich Von Roman, Rudolf Knorr, Claudia Behringer, Jakob Ruhdorfer: Brominative Deoxygenation of some aldehydes and ethers. In: Journal für Praktische Chemie/Chemiker-Zeitung. Band336, Nr.3, 1994, S.260–262, doi:10.1002/prac.19943360313 (englisch, Online [abgerufen am 6. März 2026]).
- ↑ Jonathan Clayden, Nick Greeves, Stuart G. Warren: Organic chemistry. 2nd ed Auflage. Oxford University Press, Oxford; New YorK 2012, ISBN 978-0-19-927029-3, S.127–128 (englisch).
- ↑ Robert J. H. Gregory: Cyanohydrins in Nature and the Laboratory: Biology, Preparations, and Synthetic Applications. In: Chemical Reviews. Band99, Nr.12, 8. Dezember 1999, S.3649–3682, doi:10.1021/cr9902906 (englisch).
- ↑ Hiroshi Ohno, Atsunori Mori, Shohei Inoue: Lanthanoid(III) Alkoxides as Novel Catalysts for a Rapid Transhydrocyanation from Acetone Cyanohydrin to Aldehydes and Ketones. In: Chemistry Letters. Band22, Nr.2, 1. Februar 1993, S.375–378, doi:10.1246/cl.1993.375 (englisch).
- ↑ Wayiza Masamba: Petasis vs. Strecker Amino Acid Synthesis: Convergence, Divergence and Opportunities in Organic Synthesis. In: Molecules. Band26, Nr.6, 18. März 2021, S.1707, doi:10.3390/molecules26061707, PMID 33803879, PMC 8003338 (freier Volltext) – (englisch).
- ↑ N-Heterocyclic carbenes in synthesis. Wiley-VCH; John Wiley [distributor], Weinheim: Chichester 2006, ISBN 3-527-31400-8, S.281–282 (englisch).
- ↑ David Kieslich, Jens Christoffers: Cyanide Anions as Nucleophilic Catalysts in Organic Synthesis. In: Synthesis. Band53, Nr.19, Oktober 2021, S.3485–3496, doi:10.1055/a-1499-8943 (englisch).
- ↑ Acetaldehyde Market Size, Share & Growth Report [Latest]. Abgerufen am 16. Februar 2024 (englisch).
- ↑ M. S. Peters, J. A. Quinn: Pentaerythritol Production Yields. In: Industrial & Engineering Chemistry. Band47, Nr.9, September 1955, S.1710–1713, doi:10.1021/ie50549a016 (englisch).
- ↑ David E. Lewis: Aleksei Yevgen'evich Chichibabin (1871–1945): A Century of Pyridine Chemistry. In: Angewandte Chemie International Edition. Band56, Nr.33, 7. August 2017, S.9660–9668, doi:10.1002/anie.201611724 (englisch).
- ↑ a b c Adam W. Franz, Helmut Kronemayer, Daniel Pfeiffer, Roman D. Pilz, Günther Reuss, Walter Disteldorf, Armin Otto Gamer, Albrecht Hilt: Formaldehyde. In: Ullmann's Encyclopedia of Industrial Chemistry. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany 2016, ISBN 978-3-527-30673-2, S.1–34, doi:10.1002/14356007.a11_619.pub2 (englisch).
- ↑ a b c Butyraldehyde Market Size, Share, Growth & Forecast, 2032. Abgerufen am 15. Februar 2024 (englisch).
- ↑ N-Butanol Market Size, Growth, Analysis & Forecast, 2032. Abgerufen am 15. Februar 2024 (englisch).
- ↑ 2-Ethylhexanol Market Size, Growth, Industry Report. Abgerufen am 15. Februar 2024 (englisch).
- ↑ Sandro Nalli, Owen J. Horn, Adam R. Grochowalski, David G. Cooper, Jim A. Nicell: Origin of 2-ethylhexanol as a VOC. In: Environmental Pollution. Band140, Nr.1, März 2006, S.181–185, doi:10.1016/j.envpol.2005.06.018 (englisch).
- ↑ Hydrogen Cyanide. In: Synthetic Nitrogen Products. Kluwer Academic Publishers, Boston 2005, ISBN 0-306-48225-8, S.347–360, doi:10.1007/0-306-48639-3_19 (englisch).
- ↑ Thomas Willke: Methionine production—a critical review. In: Applied Microbiology and Biotechnology. Band98, Nr.24, Dezember 2014, S.9893–9914, doi:10.1007/s00253-014-6156-y (englisch).
- ↑ Formaldehyde Market Size, Growth, Analysis & Forecast, 2032. Abgerufen am 15. Februar 2024 (englisch).
- ↑ A Nuryawan, I Risnasari, T Sucipto, A Heri Iswanto, R Rosmala Dewi: Urea-formaldehyde resins: production, application, and testing. In: IOP Conference Series: Materials Science and Engineering. Band223, Juli 2017, S.012053, doi:10.1088/1757-899X/223/1/012053 (englisch).
- ↑ Christian Carrot, Amine Bendaoud, Caroline Pillon: Polyvinyl Butyral. In: Plastics Engineering. CRC Press, 2015, ISBN 978-1-4665-7722-0, S.89–137 (englisch).
- ↑ Alexander T. Cartus, Dirk W. Lachenmeier, Sabine Guth, Angelika Roth, Matthias Baum, Patrick Diel, Gerhard Eisenbrand, Barbara Engeli, Michael Hellwig, Hans‐Ulrich Humpf, Hans‐Georg Joost, Sabine E. Kulling, Alfonso Lampen, Doris Marko, Pablo Steinberg, Wim Wätjen, Jan G. Hengstler, Angela Mally: Acetaldehyde as a Food Flavoring Substance: Aspects of Risk Assessment. In: Molecular Nutrition & Food Research. Band67, Nr.23, Dezember 2023, doi:10.1002/mnfr.202200661 (englisch).
- ↑ Ram S. Verma, Rajendra C. Padalia, Ved R. Singh, Prakash Goswami, Amit Chauhan, Balakishan Bhukya: Natural benzaldehyde from Prunus persica (L.) Batsch. In: International Journal of Food Properties. 9. Juni 2017, ISSN 1094-2912, S.1–5, doi:10.1080/10942912.2017.1338728 (tandfonline.com [abgerufen am 26. März 2026]).
- ↑ U. Krings, R. G. Berger: Biotechnological production of flavours and fragrances. In: Applied Microbiology and Biotechnology. Band49, Nr.1, 26. Januar 1998, ISSN 0175-7598, S.1–8, doi:10.1007/s002530051129 (springer.com [abgerufen am 29. März 2026]).
- ↑ Werner Köhler, Rainer Ansorg: Medizinische Mikrobiologie. Elsevier, Urban & Fischer Verlag, 2001, ISBN 3-437-41640-5, S. 92.
- ↑ Anton C. De Groot, Margo Veenstra: Formaldehyde‐releasers in cosmetics in the USA and in Europe. In: Contact Dermatitis. Band62, Nr.4, April 2010, S.221–224, doi:10.1111/j.1600-0536.2009.01623.x (englisch).
- ↑ Rooban Thavarajah, VidyaKazhiyur Mudimbaimannar, Joshua Elizabeth, UmadeviKrishnamohan Rao, Kannan Ranganathan: Chemical and physical basics of routine formaldehyde fixation. In: Journal of Oral and Maxillofacial Pathology. Band16, Nr.3, 2012, S.400, doi:10.4103/0973-029X.102496, PMID 23248474, PMC 3519217 (freier Volltext) – (englisch).
- ↑ Stefanie Federle, Stefanie Hergesell, Sebastian Schubert: Die Stoffklassen der organischen Chemie. Springer Berlin Heidelberg, 2017, ISBN 978-3-662-54968-1, S.186 (eingeschränkte Vorschau in der Google-Buchsuche).
- ↑ Ludwig Acker, Gerhard Bressau, Georg Benedikt Brubacher, Karl Maximilian Bürger, Stefan Diemair, Willibald Diemair, Klaus Doerffel, Rudi Franck, Hansfriedel Gudjons, Paul Joppien, Ludwig Kotter, Ernst Kröller, Hermvn Libert, Helmut Mühlschlegel, Titus Niedermaier, Konrad Pfeilsticker, Gerhard Pfleiderer, Wilhelm Postel, Hanspeter P. Probst, W. Rödder, Werner Schäfer, Leopold Schmid, Erich Schneider, Artur Seher, Hans Sommer, Helmut Thaler, Jean Paul Vuilleumier, Herbert Woidich: Analytik der Lebensmittel Nachweis und Bestimmung von Lebensmittel-Inhaltsstoffen. Springer-Verlag, 2013, ISBN 978-3-642-46069-2, S.648 (books.google.com).