
{kind=link}
{kind=link}
Abstract
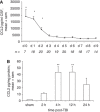
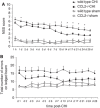
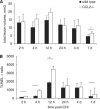
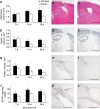
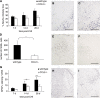
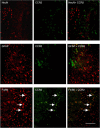
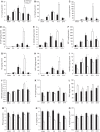
Cerebral inflammation involves molecular cascades contributing to progressive damage after traumatic brain injury (TBI). The chemokine CC ligand-2 (CCL2) (formerly monocyte chemoattractant protein-1, MCP-1) is implicated in macrophage recruitment into damaged parenchyma after TBI. This study analyzed the presence of CCL2 in human TBI, and further investigated the role of CCL2 in physiological and cellular mechanisms of secondary brain damage after TBI. Sustained elevation of CCL2 was detected in the cerebrospinal fluid (CSF) of severe TBI patients for 10 days after trauma, and in cortical homogenates of C57Bl/6 mice, peaking at 4 to 12 h after closed head injury (CHI). Neurological outcome, lesion volume, macrophage/microglia infiltration, astrogliosis, and the cerebral cytokine network were thus examined in CCL2-deficient (-/-) mice subjected to CHI. We found that CCL2-/- mice showed altered production of multiple cytokines acutely (2 to 24 h); however, this did not affect lesion size or cell death within the first week after CHI. In contrast, by 2 and 4 weeks, a delayed reduction in lesion volume, macrophage accumulation, and astrogliosis were observed in the injured cortex and ipsilateral thalamus of CCL2-/- mice, corresponding to improved functional recovery as compared with wild-type mice after CHI. Our findings confirm the significant role of CCL2 in mediating post-traumatic secondary brain damage.
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
-
- Banisadr G, Queraud-Lesaux F, Boutterin MC, Pelaprat D, Zalc B, Rostene W, Haour F, Parsadaniantz SM. Distribution, cellular localisation and functional role of CCR2 chemokine receptors in adult rat brain. J Neurochem. 2002;81:257–269. - PubMed
-
- Beni-Adani L, Gozes I, Cohen Y, Assaf Y, Steingart RA, Brenneman DE, Eizenberg O, Trembolver V, Shohami E. A peptide derived from activity-dependent neuroprotective protein (ADNP) ameliorates injury response in closed head injury in mice. J Pharmacol Exp Ther. 2001;296:57–63. - PubMed
-
- Berman JW, Guida MP, Warren J, Amat J, Brosnan CF. Localisation of monocyte chemoattractant peptide-1 expression in the central nervous system in experimental autoimmune encephalomyelitis and trauma in the rat. J Immunol. 1996;156:3017–3023. - PubMed
-
- Buttram SD, Wisniewski SR, Jackson EK, Adelson PD, Feldman K, Bayir H, Berger RP, Clark RS, Kochanek PM. Multiplex assessment of cytokine and chemokine levels in cerebrospinal fluid following severe pediatric traumatic brain injury: effects of moderate hypothermia. J Neurotrauma. 2007;24:1707–1717. - PubMed
-
- Bye N, Habgood MD, Callaway JK, Malakooti N, Potter A, Kossmann T, Morganti-Kossmann MC. Transient neuroprotection by minocycline following traumatic brain injury is associated with attenuated microglial activation but no changes in cell apoptosis or neutrophil infiltration. Exp Neurol. 2007;204:220–233. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
{kind=link}